journal menu
journal menu




issue contents
December 1998 issue
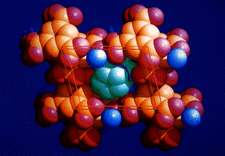
Cover illustration: Space filling representation of the hydrated anionic framework formed by trimesic acid around piles of cobalticinium cations in crystalline [(C5H5)2Co][(H3TMA)(H2TMA)].2H2O. Graphics (picture or drawing) obtained using SCHAKAL by E. Keller, University of Freiburg, Germany. Courtesy of D. Braga and F. Grepioni
research papers



Download citation





Download citation

ZrV2O7 and related phases are of interest for their negative thermal expansion properties, but there has been no detailed report of their structures to date. We describe here the room-temperature 3 × 3 × 3 superstructure of ZrV2O7, which has been determined from single-crystal synchrotron X-ray data.

Single crystals with the composition Mg0.4Al2.4O4 show a pressure-induced phase transition at 10.7 GPa which is partially reversible on pressure release. The resulting compound reveals a disordering of the cations on the tetrahedral and octahedral sites of the spinel structure.
Download citation
Download citation

Pentatitanium tetraoxide tetrakis(phosphate) belongs to the Fe2O(PO4) structure family. The relationship is indicated by the formula □3Ti5O4(PO4)4. X-ray single-crystal and high-resolution transmission electron microscopy investigations reveal partial disorder of Ti4+ over eight available octahedral voids. The ideal structure consists of four (TiO)O5, one TiO6 and four slightly distorted PO4 units.

The cation arrays in rare earth oxides reproduce the topology and distances of the structures of the pure metals.

An interatomic potential is developed for a static and vibrational simulation of α-Al2O3. Structural, elastic and thermal expansion data are predicted in the p,T domain from 0 to 40 GPa and from 300 to 1700 K.
Download citation
Download citation

The atomic structure of lead zirconate, PbZrO3, and its variation with temperature were determined by pulsed neutron diffraction using both Rietveld refinement and atomic pair distribution function analysis. The symmetry at low temperatures was shown to be Pbam and a new antiferroelectric structure was identified for the intermediate phase.

Use of the centroid concept for coordination polyhedra enables comparison of distorted and ideal polyhedra with the same number of coordinated atoms. The relation between the volumes of the distorted and the ideal polyhedra can be used as a global measure of distortion.
Download citation
Download citation

The multipole deformation electron density, Laplacian of the electron density and (3,-1) bond critical point parameters are analyzed in topaz, Al2[SiO4]F2, using accurate X-ray diffraction data.

Simple tilting of the octahedral units in perovskite gives rise to a number of distinct, closely related structures. Fifteen possible space groups are listed along with group–subgroup relationships and an indication of which translations between the different structures are allowed to be continuous.
Download citation
Download citation

The phase transitions observed in TlH2PO4 and TlD2PO4 with decreasing temperature are analysed.

The anisotropy of structural distortion of the title compound under pressure was shown to be qualitatively different to that on cooling. The role of non-covalent interactions, in particular hydrogen bonds, in the anisotropy of structural distortion is discussed.
Download citation
Download citation

The structures of the fluoroarsenates KAsF6-n(OH)n, n = 0, 1, 2, depend on the number and type of hydrogen bonds; a standard structure, a superstructure and an incommensurate modulated structure are found for n = 0, 1 and 2, respectively. A heavy-atom method developed for modulated structures is used for the solution of the incommensurate structure.
Download citation
Download citation

The Si-O and Al-O bonds in a natural zeolite, scolecite, are characterized by the topological features of the experimental electron density. The estimation of the atomic net charges in this compound is described according to data analysis by the statistical prediction matrix (leverages) method.
Download citation
Download citation

Subcell and superstructures of Er10Ru10C19 are described where various stacking variants arise from an ordered arrangement of single C atoms and C2 pairs which are situated in trigonal Ru prisms. The Ru and C atoms of the three-dimensionally infinite polyanionic network seem to obey the 18- and 8-electron rules, respectively.
Download citation
Download citation

The standard error on an intensity can be reduced below counting statistical error margins when, in a fixed time experiment, a repetition of fast profile scans replaces the traditional slow, but single scan. The decrease in intensity errors is not followed by a similar reduction in parameter errors, because the latter are dictated by the randomization of systematic intensity errors.
CCDC reference: 132038

The crystallographic and spectroscopic analyses of 2,3-diketopiperazine are combined with ab initio calculations for both the gas phase and solid state. The calculated IR shifts for the C=O stretching frequencies, which are hydrogen-bonding dependent, fit with the experimental evidence.
Download citation
Download citation

A statistical analysis of hydrogen-bond interactions formed by oximes and carboxylic acids is presented. Ab initio quantum-chemical calculations have been used to rationalize the observed preference for the oxime–carboxyl interaction compared with carboxyl–carboxyl and oxime–oxime interactions.
CCDC reference: 132035
Download citation
Download citation

Packing potential energy calculations for host–guest model crystals account for the disorder of ethyl acetate solvent molecules in the crystal state.
CCDC reference: 132039
Download citation
Download citation

Symmetry analysis of numerous triclinic structures retrieved from the Cambridge Structural Database with P-1 and Z = 8 revealed a rich variety of *21/*c pseudosymmetry, which may exist in the oblique unit cells. The migrations of these symmetries are accompanied by the irregular translations of their complementary operators, which may lead even to transitional space-group formations.
CCDC reference: 132037
Download citation
Download citation

The temperature dependence of the geometries of the hydrogen bonds before and after the second-order phase transition (Tc = 170 K) is studied. The O⋯O distances of two O-H⋯O hydrogen bonds involving the hydrogen succinate ions increase as the temperature decreases below Tc and the N⋯O distance of a weak bifurcated hydrogen bond involving the ammonium ion decreases below Tc.
Download citation
Download citation

Crystal structures of eleven 2,4,6-triisopropylbenzophenones have been determined to show that the molecular structures are essentially the same, independent of the substituted groups. The photostability of 4'-COOMe and 4'-COCl derivatives in the solid state is attributed to the narrow reaction cavity around the triisopropylphenylcarbonyl moiety.
Download citation
Download citation

The diastereoselective photocyclization of the title compound was directly observed as a crystal-to-crystal transformation. The crystal structure after 100% conversion has been determined, revealing that the carbonyl C and the isopropyl central C atoms approach each other to form a C-C bond.
Download citation
Download citation

Experimentally determined properties of 2-pyridone include the total electron density and its Laplacian, the electrostatic potential and the dipole moment. These are used to characterize the N-H⋯O, C-H⋯O and C-H⋯π interactions.
CCDC reference: 132036

Nearly 200 examples of local centers of symmetry have been found in each of the space groups Pca21 and Pna21 for Z > 4. These are concentrated about x=1/8, y=1/4 and x=1/8, y=0, respectively. Implications for the refinement process are discussed.
short communications
Download citation
Download citation

The original refinement of this structure, in space group P63, was restrained so as to yield a presumed composition of approximately Al76Cr18Ni6. Revision of the space group, to P63/m, permits unrestrained refinement which indicates no more than a trace amount of Ni.
books received
Free 

international union of crystallography
Free