journal menu
journal menu





issue contents
January 1999 issue
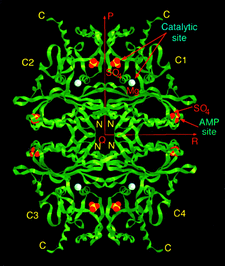
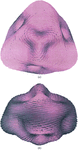
Cover illustration: A ribbon diagram of rabbit liver fructose-1,6-bisphosphatase homotetramer, showing bound magnesium and sulfate ions at catalytic and AMP binding sites (p. 93).
editorial




Free 
letters to the editor
Free

The background to the deposition and release of macromolecular structural data and the main issues to consider are discussed here and some recommendations are made.
Free

Deposition of structure-factor data with the PDB is described.
fast communications

Post-crystallization soaking of MTCP-1 crystals in 2.0 M ammonium sulfate instead of the 1.5 M ammonium sulfate used for crystallization improved the diffraction from 3.0 to 2.0 Å resolution.
research papers



The three-dimensional structure of carbamoyl phosphate synthetase from Escherichia coli has been determined and refined to 2.1 Å resolution. This high-resolution structural analysis has allowed for a complete description of the active sites associated with both the small and large subunits.
PDB reference: carbamoyl phosphate synthetase, 1jdb
Structure of the bifunctional inhibitor of trypsin and α-amylase from ragi seeds at 2.9 Å resolution
The bifunctional inhibitor of trypsin and α-amylase has been purified from seeds of ragi. The three-dimensional structure has been determined by X-ray diffraction.
PDB reference: trypsin inhibitor, 1b1u
The crystal structural analysis of bovine heart cytochrome c oxidase has been determined at 2.8 Å resolution by the multiple isomorphous replacement (MIR) method with three heavy-atom derivatives. Out of 3606 amino-acid residues in 26 subunits of a dimeric enzyme, 3560 amino-acid residues were located in the electron-density map calculated with MIR phases.
PDB reference: bovine heart cytochrome c oxidase, 1occ
X-ray structure of the orthorhombic form of bovine phospholipase A2 has been refined at 1.5 Å resolution with an ordered surface loop.
PDB reference: bovine pancreatic phospholipase A2, 1une
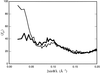
A novel single-crystal neutron diffractometer utilizing a polychromatic neutron beam reduces data-collection times by about one order of magnitude. It is specifically suited for applications involving crystals of biological macromolecules. It was used to determine the arrangement of solvent molecules in the crystal structure of coenzyme B12r.
Two complexes between rainbow trout lysozyme (RBTL) and 4-methylumbelliferyl chitobioside, 4MeU-(GlcNAc)2, and chitotrioside, 4MeU-(GlcNAc)3, have been produced by co-crystallization and soaking, respectively, and the crystal structures have been solved at 2.0 Å resolution.
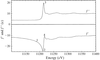
A multiple wavelength anomalous diffraction (MAD) experiment was performed on an iridium derivative of hen egg-white lysozyme. The results show how systematic error in MAD data due to sample absorption can be reduced where a white line is present in the absorption spectrum of a heavy atom.

The crystal structure of rice dwarf virus was determined with ab initio phasing up to 20 Å resolution. The electron-density map shows the RNA shell inside the particle as well as double-shell structure consisting of proteins.

The crystal structure of the C-terminal domain of the HIV-1 capsid protein has been refined at 2.6 Å resolution. The protein crystallizes as a dimer, and the structure reveals details of the essential capsid protein dimer interface including salt bridges, hydrogen bonds and van der Waals contacts.
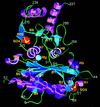
The crystal structure of the R-form of rabbit liver fructose-1,6-bisphosphatase at 2.3 Å resolution shows that it is homologous to the enzyme from pig kidney. An indogenous Mg2+ ion is bound at the metal binding site of the catalytic cleft.
PDB reference: fructose 1,6-bisphosphatase, 1bk4
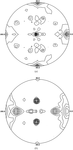
BPTI crystallizes from thiocyanate as a decamer. The ten molecules aggregate as a globular micelle-like particle with strong internal contacts.
PDB reference: bovine pancreatic trypsin inhibitor, 1bhc
The crystal structure of orthorhombic hen egg-white lysozyme has been refined at 1.7 Å resolution. The correlation between crystal forms and intermolecular macrobonds A, B and C were evaluated semi-quantitatively.
PDB reference: orthorhombic lysozyme, 1bgi
The CDRH2 of the 2E8 antibody to the low-density lipoprotein (LDL) receptor binding region of apolipoprotein E (apoE) mimics the LDL-receptor repeat 5. The 2E8 structure therefore provides an initial model for the electrostatically dominated apoE–LDL receptor interaction.
PDB reference: 2E8 Fab antibody fragment, 12e8
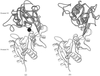
The method of molecular replacement is used to determine the structure of a merohedrally twinned protein crystal.
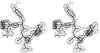
The primary and secondary binding of two similar trypsin inhibitors from squash seeds in complex with bovine and anionic salmon trypsins are discussed in order to find explanations for substrate discrimination in trypsins.

A crystallogenesis study including a two-dimensional phase diagram yielded two crystal forms of N-terminal truncated yeast aspartyl-tRNA synthetase suitable for structure determination.
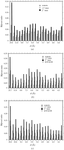
Moduli of envelope structure factors have been extracted from MASC data for three proteins of very different molecular weights, and they are compared with values obtained from model envelopes. The existence of ordered anomalous sites and their MAD effects are shown in all three cases.

A known small protein, RNAP1, with more than 800 non-H atoms, none heavier than sulfur, has been solved with MULTAN88. Objective procedures gave a detailed structure showing all atoms starting from 1.17 Å resolution data.
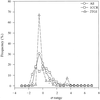
The correlation coefficients between the mean atomic displacement parameters (B values) of the main-chain and the side-chain atoms in high-resolution protein structures are distributed over a broad range related to the refinement package used. The frequency distribution of B values at Cα atoms could possibly be developed as a qualitative validation tool.

Homologous models of penicillopepsin were used to generate initial phases for the native crystal structure of the aspartic proteinase penicillopepsin by molecular replacement. It is shown that the combination of cross-validation, torsion-angle dynamics simulated annealing and maximum-likelihood target functions allow refinement to proceed efficiently, even when the initial molecular-replacement-phased electron-density maps do not allow manual rebuilding.
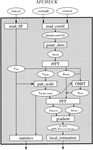
SFCHECK collates a number of objective criteria for evaluating the quality of the X-ray data and for assessing the agreement of the model with those data. It is geared to analyze protein and nucleic acid crystal structures, which also contain water and other small molecules.
A difference electron-density map in which water molecules are deliberately removed from the structure-factor calculation contains a wealth of information concerning the correctness of a crystal structure. The program DDQ uses this information in an automated fashion to arrive at local and global indictations for the accuracy of crystal structures.
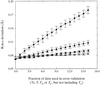
More precise structures are possible through improved free R factors. Changed scaling procedures allowing reduction in the size of the validation set and use of approximations to the validation amplitudes lead to more accurate electron-density maps. Improvements are greatest for real-space refinement, but are also applicable to model building and phase refinement.
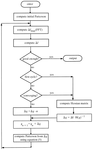
Proper NCS rotation axes can be identified and located in surprisingly poor electron-density maps. The described program performs an overall search using reasonable computing time.
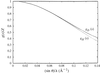
Globbic scattering factors have been calculated for main-chain peptide units and amino-acid side-chain groups to 3 Å resolution via Debye's formula. They are also approximated by a single-Gaussian formula.
A maximum-entropy algorithm for estimating reflection intensities of unobserved Laue diffraction spots is described. Test results have shown that this method is capable of producing useful data to fill in the `low-resolution hole'.
The low-resolution molecular shape of nitrite reductase (NiR), determined from solution X-ray scattering data, was located in the crystallographic unit cell by a molecular-search method. Good low-resolution phases were obtained from which phase extension could be attempted.
Individual anisotropic refinement of atomic thermal parameters using FFT opens new perspectives for the reparametrization of macromolecular models.
crystallization papers

Single crystals of the 1:1 complex of colicin E9 DNAase domain and its cognate immunity protein Im9 have been grown. They belong to space group P212121 and diffract to 2.15 Å resolution. Crystals of selenomethionine-substituted protein have also been prepared for MAD phasing experiments.
A truncated soluble form of the murine class I major histocompatibility antigen complex H–2Dd was cloned, expressed, refolded in vitro and crystallized in a complex with murine β2 microglobulin and the peptide RGPGRAFVTI from the V3-loop of the gp160 HIV-1 protein. Crystals belonging to the space group P212121 with cell dimensions a = 51.3, b = 92.5, c = 108.8 Å were obtained.
RuvA has been overexpressed in E. coli, purified and cocrystallized with a synthetic Holliday junction substrate made from four 18-base deoxyoligonucleotides. Crystals grown using the hanging-drop vapour-diffusion method with sodium acetate as the precipitant diffract to 6 Å and belong space group C2, with cell parameters a = 148, b = 148, c = 106 Å and β = 123°.
This paper describes the purification, crystallization and initial X-ray analysis of the carotenoid-binding protein, V600, of the chondrophore V. velella.

Crystals of a novel phospho-histidine phosphatase SixA for the histidine-containing phosphotransfer (HPt) domain of the E. coli sensor kinase ArcB were obtained. The crystals belong to space group P212121 (a = 39.26, b = 48.62 and c = 83.18 Å) and diffract up to 1.5 Å resolution using synchrotron radiation.

Preliminary crystallographic analysis of a guanine-exchange factor for small GTP-binding proteins of the ras family
A macromolecular complex of human transthyretin, human retinol-binding protein and an anti-retinol-binding protein Fab has been crystallized. Crystals diffracted to a resolution limit of 3.36 Å using synchrotron radiation.
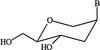
Crystallization and preliminary X-ray characterization of the strong hybridizing hexitol nucleic acid is described.
A 23-mer RNA corresponding to the dimerization-initiation site of HIV-1 RNA has been crystallized. Crystals are sometimes twinned by hemihedry and the corresponding X-ray data require specific treatment prior to be used.

The ATP-binding-cassette (ABC) protein MalK is the energy-providing component of an ABC transport complex which translocates maltose and maltodextrin across the membrane. The obtained crystals of the MalK protein diffract to about 3.0 Å resolution.

Pyrimidine nucleoside phosphorylase from Bacillus stearothermophilus has been cloned, purified and crystallized in space group P21 (diffraction to 2.7 Å) in the presence of the substrate-analog inhibitor pseudouridine.
YPD1 has been expressed in Escherichia coli, purified to homogeneity and crystallized. The crystals were obtained by hanging-drop vapor-diffusion using PEG 4000 as a precipitant. Preliminary X-ray diffraction analysis indicates that the crystals belong to tetragonal space group P43212 or P41212 with unit-cell dimensions a = b = 52.71, c = 244.02 Å.
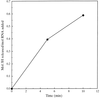
Xylose isomerase from T. caldophilus shows the maximum activity at 363 K. It has been crystallized into an orthorhombic space group P212121.
NADH oxidase from T. aquaticus has been crystallized and the crystals have been characterized using synchrotron radiation. They belong to space group P622 with unit-cell dimensions a = b = 89.9 and c = 491.6 Å.

The gene encoding Campylobacter jejuni ferritin, a prokaryotic ferritin, has been overexpressed and the protein purified. Preliminary crystallization and diffraction studies show the crystal belongs to the I432 space group (a = 151.52 Å). The structure solution by molecular replacement is in progress.
Histidine-tagged RluC was cloned, overexpressed and purified by nickel-affinity chromatography. Crystals of a proteolytically derived fragment, grown by precipitation with sodium acetate at pH 8.0, belong to space group P321, with unit-cell dimensions a = b = 97.1, c = 86.3 Å and have two molecules in the crystallographic asymmetric unit.

Anthranilate synthase catalyzes the first step in the biosynthesis of tryptophan from chorismate. The tryptophan-inhibited form of the partial complex from S. typhimurium has been crystallized in space group P21212 with dimensions a = 116.7, b = 101.2 and c = 66.8 Å.

Iso-2 azurin, which functions as an electron acceptor for methylamine dehydrogenase, has been crystallized using two kinds of precipitants: PEG 4000 and ammonium sulfate. Preliminary data for the crystals obtained are presented in this paper.

The crystal structure of the L8S8 rubisco from A. eutrophus has been solved to 3.2 Å resolution. A 35 resolution extension in the small subunits forms a structural motif in which antiparallel strands from four subunits contribute to a new eight-stranded β-barrel.

A domain derived from streptococcal protein G has been used to obtain crystals of an antibody Fab fragment in complex with a peptide antigen from the bacterial pathogen Neisseria meningitidis.

Phospholipase D from S. antibioticus has been crystallized in six crystal forms, of which the type VI crystals are suitable for X-ray structure determination.

Crystals of E. carotovora ssp. carotovora polygalauronase have been grown. They have a single molecule in the asymmetric unit and diffract to 1.9 Å.
A biotin-binding RNA has been crystallized using the vapor-diffusion method and polyethylene glycol as precipitant. Crystals diffract to 2.8 Å and belong to space group P4222.
Affinity purification of an in vitro selected RNA yields highly diffracting crystals of an aptamer–ligand complex.

Two crystal forms of pyruvate-ferredoxin oxidoreductase from the sulfate-reducing bacteria Desulfovibrio africanus have been obtained. They belong to space group P212121 and diffract to 2.3 Å resolution using synchrotron radiation.

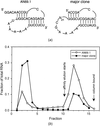
The complex formed by the E. coli methionyl-tRNA formyltransferase with its product, the formyl-methionyl initiator tRNA, has been crystallized. The crystals diffract to 2.8 Å resolution.
formyltransferase with its product, the formyl-methionyl initiator tRNA, has been crystallized. The crystals diffract to 2.8 Å resolution.
short communications

The structure of bovine pancreatic trypsin inhibitor has been solved in a new crystal form where protein molecules create a decameric assembly.
PDB reference: bovine pancreatic trypsin inhibitor, 2hex

An M. jannaschii fibrillarin homologue was overexpressed, purified and crystallized. The crystals diffract to 1.8 Å resolution.

Three bioactivity-variant neurotoxins, including an unusually acidic and a neutral components, from Chinese scorpion venom have been purified and crystallized, which provide a valuable system for studying structure–function relationships.

Crystallization of orotate phosphoribosyltransferase has been achieved at medium temperature.
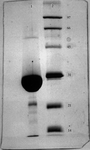
Recombinant uracil phosphoribosyltransferase (UPRT) enzyme of Toxoplasma gondii was expressed in Escherichia coli and purified from the cell-free extract by a combination of chromatographic steps. The protein was crystallized using the hanging-drop vapor-diffusion technique with ammonium phosphate as precipitant and gave unit-cell dimensions a = b = 119.9, c = 70.8 Å and two molecules per asymmetric unit.
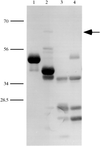
Purification and crystallization of an active serpin fromthe parasite S. haematobium, a membrane-anchored protein, is reported. The crystals belong tothe trigonal space group P3221 or P3121 with a = b = 64.7, c = 186.7 Å. α = 90.0, β = 90.0 and γ = 120.0° and diffracted to 2.2 Å.
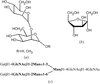
An efficient method for obtaining co-crystals of the plant lectin concanavalin A is described. This rapid crystallization and structure determination is required if crystallography is to impact on other biophysical techniques.

This paper presents the expression, purification and crystallization of a BH (GTPase activating) domain from a GTPase regulatory protein associated with focal adhesion kinase. Native crystals diffract to 2.15 Å and a full data set has been collected.
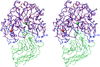
Comparisons between the interaction patterns of a bean inhibitor with mammalian and insect α-amylases provide insights about the main rules underlying the inhibition exerted by the proteinaceous inhibitor.
international union of crystallography
Free