journal menu
journal menu





issue contents
September 1999 issue
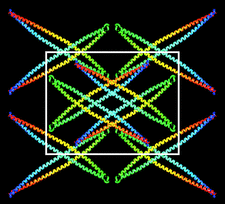
Cover illustration: Molecular-packing model, shown projected along the a axis, for an apo ![[Delta]](/logos/entities/Delta_rmgif.gif) (1-43)A-I extended helical rod. Each monomer is coloured from red at the N-terminus to blue at the C-terminus (p. 1578).
(1-43)A-I extended helical rod. Each monomer is coloured from red at the N-terminus to blue at the C-terminus (p. 1578).
fast communications




Crystals of model protein thaumatin grown within agarose gel in microgravity are shown to be of better crystallographic quality than controls prepared in parallel on earth.
research papers


The crystal structure of the decamer d(GGCCAATTGG) at atomic resolution (1.15 Å) provides detailed information about B-DNA and triplex hydration.


The structure of the RhoA–RhoGDI complex has been investigated by X-ray diffraction at 4.0 Å resolution, using experimental phasing and molecular replacement. The globular C-terminal domain of RhoGDI is bound to an epitope on RhoA which includes residues at the base of the switch II region and in the neighboring helix α3, while the flexible N-terminal fragment occludes the Dbl-like GEF-binding epitope.
PDB reference: RhoA–RhoGDI complex, 1cc0
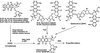
Epidoxorubicin covalently binds d(CGCGCG) from its 3′-amino substituent to a 2-amino group of a G base via Schiff-base chemistry with formaldehyde. Covalent attachment coupled with non-covalent interactions function to virtually crosslink the model DNA. Cell experiments suggest similar binding in the nuclear DNA of cancer cells.
PDB reference: epidoxorubicin–CH2–d(CGCGCG), 1qda

A mutant of human insulin (B9 Ser→Glu) has been crystallized and its structure determined to 2.5 Å resolution.
PDB reference: Human insulin mutant (SerB9Glu), 1b9e

A variant of E. coli thioredoxin with active-site sequence Cys32-Val33-Trp34-Cys5 complements Saccharomyces cerevisiae deficient in protein-disulfide isomerase. The 2.2 Å crystalline structure of the variant provides insight into its function.
PDB reference: CVWC thioredoxin, 1txx
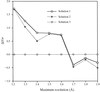
The use of molecular replacement to solve an intermediate-sized (16-residue) polypeptide, antiamoebin I, is described. The parameters and model features necessary to effect the solution are discussed.
PDB reference: antiamoebin I, 1joh

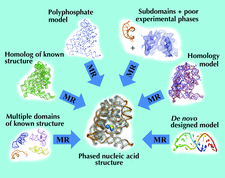
This paper shows how the structure of the large protein pyruvate:ferredoxin oxidoreductase was solved. Starting from MAD phase information in one crystal form, another form was solved by molecular replacement, using an electron-density search model improved by solvent flattening. MIR phase information was exploited in this other form and finally density-averaging methods were used for phase improvement and extension to high resolution.
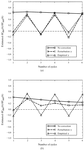
An investigation into the problem of obtaining reliable estimates of the quality of phases obtained from arbitrary density-modification techniques is presented.
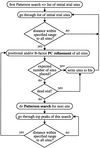
A new highly automated heavy-atom search procedure combines a fast Fourier transform translation function, Patterson superposition functions and Patterson correlation refinement. The search procedure can be applied to various native and difference Patterson maps and their statistically weighted averages.
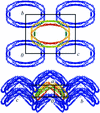
A new crystal form of apo A-I, the major protein component of high-density lipoprotein, is described. Packing arguments are presented to suggest that in this crystal form apo A-I adopts a conformation distinct from that observed in the first apo A-I crystal structure.
crystallization papers

Preliminary crystal data have been obtained for the first time for a monomeric isocitrate dehydrogenase. The enzyme crystallizes in space group C2 with a = 137.1, b = 54.6, c = 126.4 Å, β = 108.2°.
3-deoxy-D-arabino-heptulosonate-7-phosphate synthase from S. cerevisiae has been crystallized. The triclinic crystals diffract to 2.3 Å resolution.

The hanging-drop technique has been used to grow crystals of P. ostreatus lectin. Hexagonal bipyramidal crystals grew to maximum dimensions of 0.2 × 0.2 × 0.5 mm in two to three weeks.

Catabolic OTCase from P. aeruginosa has been crystallized in three crystal forms. Only cubic crystals diffract to 2.5 Å resolution.

The HMG-box domain of HMG-D has been co-crystallized with linear duplex DNA in the space group P212121 with unit-cell dimensions a = 43.74, b = 53.80, c = 86.84 Å; data have been collected to 2.20 Å resolution and multiple co-crystals with halogenated DNA have been obtained.

The expression, crystallization and preliminary X-ray analysis of the two amino-terminal immunoglobulin domains of the neural cell adhesion molecule is reported. The crystals diffract to 1.85 Å resolution and belong to space group P21. Four molecules are present in the asymmetric unit.
The N-terminal portion of the type 1 ryanodine receptor D2 region, critical for excitation–contraction coupling in skeletal muscle, has been crystallized as a glutathione S-transferase fusion protein. Using a newly developed cryo-soaking method and synchrotron radiation, X-ray diffraction data to 2.2 Å resolution have been collected.

Crystals of the restriction endonuclease EcoRII have been grown which have the symmetry of space group I23 or I213, with a unit-cell axis of 160.3 Å, and diffract to about 4 Å on a rotating-anode X-ray source.

Undecaprenyl diphosphate synthase, a Z-type prenyltransferase, from M. luteus has been crystallized. The crystals diffract to at least 2.2 Å resolution and belong to the monoclinic space group C2.

Pteridine reductase is a validated target for a structure-based approach to identify new therapies against trypanosomatid infections. Well ordered crystyals have been obtained of the L. major enzyme.

The Kunitz-type trypsin inhibitor from seeds of Flamboyant (D. regia) has been purified to homogeneity and plate-like crystals suitable for X-ray analysis have been grown by the hanging-drop method using PEG 6000 as a precipitant.

Crystallization and preliminary X-ray diffraction studies of human catalase, purified from haemolysate of human placenta, are reported to 1.76 Å resolution.

L-Asparaginase from E. coli has been crystallized in a new form. This compound is important as a chemotherapeutic drug in the treatment of childhood leukaemia.

The crystallization and preliminary X-ray analysis of an oxyhaemoglobin from the Brazilian wolf C. brachiurus are reported.
short communications

The results of the Sim distribution approach and MLPHARE are compared with those from direct methods.
Site-directed mutagenesis has been used to determine the efficacy of changing surface residues to improve crystal quality. Changes in amino-acid residues were found to affect crystallization dramatically and it is concluded that crystal engineering is a valuable tool in protein crystallography.

Crystal engineering has been used to produce well defined crystals of the 24 kDa fragment of DNA gyrase B from S. aureus.