journal menu
journal menu





issue contents
December 1999 issue
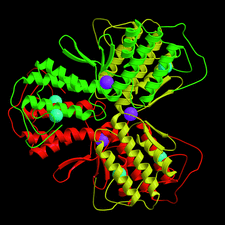
Cover illustration: Agkistrodotoxin in an orthorhombic crystal form with six molecules per asymmetric unit (p. 1986).
fast communications




Using heavily methionine-substituted T4 lysozyme as an example, it is shown how the addition or deletion of a small number of methionines can simplify the location of selenium sites for use in MAD phasing. By comparing the X-ray data for a large number of singly substituted lysozymes, it is shown that the optimal amino acid to be substituted by methionine is leucine, followed, in order of preference, by phenylalanine, isoleucine and valine.
research papers


The structure of Taq DNA polymerase in a new crystal form exhibits a significantly different orientation for the structure-specific nuclease domain from those reported. In conjunction with other data, this implies a high relative mobility between the nuclease and polymerase domains in this enzyme.
PDB reference: Taq DNA polymerase, 1cmw
Structural analysis of the Gly121Tyr dimeric form of ornithine decarboxylase from Lactobacillus 30a shows an active site similar to the dimeric units present in the native dodecamer, but with flexibility in the wing domains. A novel GTP binding model is observed and described.
PDB reference: Gly121Tyr L30a ODC, 1c4k

The crystal structure of agkistrodotoxin determined by the molecular-replacement method reveals Ca2+ ion linked dimers. Extensive intermolecular hydrophobic interactions occur at the molecular interfaces along the interfacial recognition site. The implications on the neurotoxic mechanism of agkistrodotoxin and phospholipase A2–membrane interactions are discussed.
PDB reference: agkistrodotoxin, 1bjj
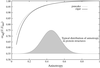
A measure Suij, expressing the similarity or dissimilarity between two sets of anisotropic displacement parameters, is easily calculated directly from the parameters Uij. This measure may be used to compare the Uij parameter estimates for independently refined protein models at near-atomic resolution. The average value of Suij per residue is evaluated for pairs of isomorphous or NCS-related protein structures taken from the Protein Data Bank.
Analysis of AA·TT dinucleotide steps in B-DNA-type oligomer crystal structures reveals that favourable inter-strand C2—H2⋯O2 interactions in the minor groove of DNA satisfy all the geometrical features required for them to be classified as C—H⋯O hydrogen bonds.

The crystallization and structure determination of apo A-I, the major protein component of high-density lipoprotein, are described. Several unusual features of the diffraction pattern are interpreted in light of the 4 Å crystal structure. Progress in assembling a 3 Å data set is reported.

The X-ray diffraction pattern from ligands aperiodically bound to a helix contains two groups of layer-lines which can be quantified by the Fourier–Bessel structure factor and the binding-distribution interference function.
crystallization papers
Recombinantly C-terminally 6-histidine-tagged E. coli argininosuccinate synthetase has been expressed, purified and crystallized. Crystals belong to space group I222, a = 79.70, b = 105.84, c = 127.33 Å, and diffract to 1.6 Å resolution.

Adenosine deaminase from bovine intestine has been crystallized. The crystals diffract to at least 2.0 Å and belong to space group P41212 or P43212.

UDP-MurNAc-tripeptide D-alanyl-D-alanine-adding enzyme (MurF) has been crystallized. The hexagonal crystals diffract to 2.8 Å resolution.

The complex between the major birch-pollen allergen Bet v 1 and a murine Fab fragment has been crystallized. Crystals diffract to 2.9 Å resolution and the complex structure is expected to serve as a starting point for identifying allergic human serum IgE-binding epitopes.

The organic solvent ethylammonium nitrate has been demonstrated to be a useful additive in protein crystallography.
Crystals of the Msx1 homeodomain–DNA complex diffracting to 2.15 Å resolution have been grown and characterized by X-ray crystallography.

Crystals of the plasmid pSM19035-encoded transcriptional repressor ω protein have been obtained in two crystal forms. X-ray diffraction data of these crystals have been collected to 2.4 and 2.9 Å resolution, respectively.
The synthesis of rhamnose is a potential therapeutic target in many bacteria. The preliminary structural study on RmlD, a key enzyme in the synthesis, is described.

Preliminary X-ray characterizations of two isoforms of yeast hexokinase are reported. Crystals of yeast hexokinase PI diffract to 3.1 Å and crystals of yeast hexokinase PII diffract to 2.2 Å.

Thermotoga maritima ribosome recycling factor has been crystallized. Diffraction data to 2.55 Å resolution have been collected.

2.9 Å resolution data have been obtained for sweet potato purple acid phosphatase. The enzyme crystallizes in space group P6522 with a = b = 118.4, c = 287.4 Å.

A laminin G-like domain of human SHBG has been overproduced and crystallized in complex with 5α-dihydrotestosterone in trigonal and tetragonal crystal forms. The trigonal crystal form diffracts better that 1.6 Å resolution and is suitable for structure determination.