journal menu
journal menu





issue contents
June 2004 issue
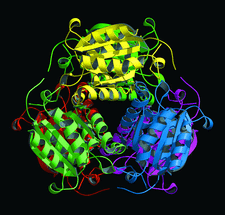
Cover illustration: Hexameric assembly of E. coli MoaB viewed along the threefold crystallographic symmetry axis (p. 1068).
research papers




The anisotropic mosaicity of protein crystals was investigated using high-resolution X-ray diffraction rocking-curve measurements. Statistical data analysis was used to quantitatively evaluate whether a homogeneous magnetic field of 2.4 T applied during crystal growth can lead to improved crystal quality in the example of the tetragonal form of hen egg-white lysozyme.



The crystal structure of A. irakense pectate lyase (PelA) has been determined at a resolution of 2.65 Å. The overall structure of PelA does not adopt the characteristic parallel β-helix fold displayed by pathogenic pectate lyases; instead, it displays a predominantly α-helical structure with irregular coils and short β-strands.
PDB reference: pectate lyase, 1r76, r1r76sf
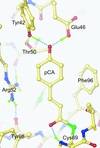
Photoactive yellow protein was solved at high resolution under a variety of conditions in order to investigate the unusually short hydrogen bonds to its coumaric acid chromophore.

A novel benzoic acid inhibitor of influenza virus neuraminidase induces a previously unobserved conformational change in the side chain of the crucial active-site residue Glu275.
PDB reference: BANA 207–B/Lee/40 NA complex, 1vcj, r1vcjsf

It is shown that in the presence of severe site-specific radiation damage the use of unmerged data and a parametrization that implements dose-dependent occupancies for the anomalous scatterers can give significant improvements in SAD/MAD and/or heavy-atom phases. Such an option is now implemented in the program SHARP; results on a brominated RNA molecule are presented.

The crystal quality of three kinds of biomolecules have been assessed by a `relative Wilson plot' method and a rational method has been developed for estimating crystal quality.

An orthorhombic actin crystal was converted into a partially hemihedrally twinned tetragonal crystal by condensation induced by flash-freezing. The structure, with two actin molecules in the crystallographic asymmetric unit, was determined by molecular-replacement methods.
PDB reference: actin, 1rfq, r1rfqsf

The crystal structure of Mn–Mn ConA grown by the gel-acupuncture method at pH 8 contains 25 molecules of glycerol, the majority of them at conserved positions. A relation exists between the crystal contacts and the space group, the contact Asp58–Ser62 being a universal feature of ConA crystals.
PDB reference: Mn–Mn ConA, 1nxd, r1nxdsf

This paper presents an efficient quaternion-based algorithm for analyzing peaks from a cross-rotation function to identify model orientations consistent with proper non-crystallographic symmetry (NCS), and to generate NCS-consistent orientations missing from the list of cross-rotation peaks.

The crystal structure of E. coli MoaB protein and a detailed comparison with the homologues MogA, Cnx1 G-domain and gephyrin are reported.
PDB reference: MoaB, 1r2k, r1r2ksf

A simple screening test for optimized stabilization and conditions for an ABC-ATPase was established that preserves activity, avoids precipitation and provided material suitable for crystallization and X-ray analysis for all of the functional states of the catalytic cycles of the HlyB-NBD.
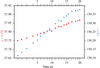
An analysis of radiation-damage-induced structural and intensity changes is presented on the model protein thaumatin.
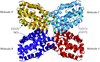
The crystal structure of PH1161 protein, a transcriptional activator B. subtilis TenA homologue from P. horikoshii, was analyzed at 2.15 Å resolution.
PDB reference: PH1161, 1udd, r1uddsf
structural genomics papers
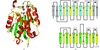
This paper reports the crystal structure of the product of the YdeN gene.
PDB reference: YdeN gene product, 1uxo, r1uxosf
crystallization papers
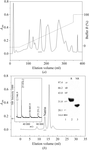
The purification and crystallization of two cysteine-rich secretory protein (CRISP) family members, natrin and stecrisp from N. atra and T. stejnegeri venoms, allowed the collection of two complete data sets to 2.1 and 1.6 Å resolution, respectively.

L. laeta SMase I, a 32 kDa sphingomyelinase present in L. laeta venom, has been crystallized and diffraction data have been collected to 1.8 Å resolution.

The DNA-binding domain of BldD from S. coelicolor has been crystallized. The crystals diffract to 1.8 Å resolution and belong to space group C2.
Nogalonic acid methyl ester cyclase, a polyketide cyclase in the biosynthetic pathway of the antibiotic nogalamycin, has been crystallized. The crystals obtained in the presence of the substrate are orthorhombic, space group I222, with unit-cell parameters a = 69.1, b = 72.0, c = 65.4 Å and diffract to 1.35 Å resolution.
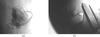
A novel plant defensin protein, SPE10, was isolated and characterized from P. erosus seeds. Preliminary crystallographic studies have been performed.
Open  access
access
access
Two peroxisomal acyl-CoA oxidase enzymes from A. thaliana have been expressed in E. coli and one of them was crystallized in a selenomethionine-substituted form. Diffraction data from both enzymes have been collected at synchrotron sources.
The cloning, overexpression and crystallization of PhzA, an enzyme involved in the final steps of phenazine-1-carboxylic acid biosynthesis, were undertaken. Details of data collection from both native and seleno-L-methionine-labelled crystals are described.

GyrA14, a fragment of GyrA, was crystallized in its free state and as a complex with the toxin CcdB.

Acylphosphatase from a hyperthermophilic archaea, P. horikoshii OT3, has been crystallized and X-ray diffraction data have been collected to 1.72 Å resolution.
Open access
access
The S. coelicolor actIII ORF5 gene has been isolated and cloned into a His-tagged vector to overexpress an active NADPH-dependent ketoreductase used in type II polyketide synthesis. Protein crystals obtained in the presence of NADP+ diffract to 2.5 Å.

A novel haemolytic lectin from the parasitic mushroom L. sulphureus has been crystallized by the hanging-drop vapour-diffusion method. Native data have been collected to 2.7 Å resolution.
The expression, purification, crystallization and preliminary X-ray analysis of a novel human DNA glycosylase are presented.
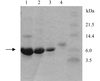
CylR2 was expressed with a C-terminal six-histidine tag and purified to homogeneity with a cobalt-affinity column followed by size-exclusion chromatography. Both native and SeMet proteins were produced and crystallized.

The thermostable endo-1,5-α-L-arabinanase ABN-TS from B. thermodenitrificans TS-3 was crystallized. A complete diffraction data set was collected to 1.9 Å resolution using synchrotron radiation.

Crystallization of B. subtilis guanine deaminase, a key enzyme in nucleotide metabolism which catalyzes the hydrolytic deamination of guanine to xanthine.
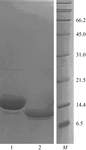
γ-Filamin is a multidomain protein present in the myofibrillar Z-disc, where it cross-links the actin filaments. Recombinant repeat 23 of γ-filamin was crystallized and a SAD data set was collected to 2.05 Å resolution.

Crystals of the motor domain of the human centromere-associated protein E (CENP-E) have been obtained by high-throughput crystallization screening using an automated TECAN crystallization robot. The best-diffracting crystals belong to space group P21, with unit-cell parameters a = 49.35, b = 83.70, c = 94.16 Å, β = 103.05°, and diffract to better than 2.1 Å.
Open access
access
Crystals of the fusion core of both Nipah and Hendra viruses were obtained; the crystals diffract X-rays to 2.0–2.1 Å.

Crystallization and preliminary X-ray crystallographic analyses of EcoO109I and its complex with DNA
Tetragonal and orthorhombic crystals of EcoO109I and its complex with DNA were obtained and diffraction data were collected to 2.4 and 1.9 Å resolution, respectively.

α-Isopropylmalate synthase from M. tuberculosis (Rv3710), which catalyses the first committed step of leucine biosynthesis, has been expressed, purified and crystallized in a form suitable for high-resolution X-ray structural analysis.
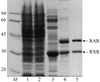
This study describes the crystallization and preliminary X-ray analysis of a heterodimer containing the ligand-binding domains of the retinoid X receptor α and of the retinoic acid receptor β in complex with 9-cis-retinoic acid and a fragment of the transcriptional coactivator TRAP220. The complex crystallizes in space group P3121 and diffracts to 2.9 Å resolution.
short communications

The crystal structure of the plant-type asparaginase from E. coli, EcAIII, has been solved by molecular replacement and refined to a resolution of 1.65 Å. Sodium ion-binding sites have been identified that were not observed in other related enzymes. The structure of EcAIII helps to explain some of the differences between the substrate specificities of plant-type asparaginases and glycosylasparaginases.
PDB reference: isoaspartyl peptidase, 1t3m, r1t3msf
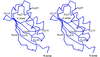
The crystal structure of a novel acylphosphatase (AcpDro2) has been solved at 1.5 Å resolution in order to improve knowledge of the enzyme catalytic cycle as well as of the molecular basis of protein folding, misfolding and aggregation.
PDB reference: AcpDro2, 1urr, r1urrsf
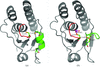
The ArsC triple mutant C10SC15AC82S has a structure that is very similar to that of the reduced form of wild-type ArsC, with a folded redox helix and a buried catalytic Cys89. In the adduct form, the TNB molecule is buried in a hydrophobic pocket and the disulfide bridge between TNB and Cys89 is sterically inaccessible to thioredoxin.
addenda and errata
Open access
access