journal menu
journal menu





issue contents
September 2004 issue
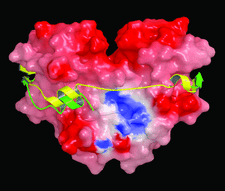
Cover illustration: YscM2 (green ribbon), a negative regulator of type III secretion in Yersinia enterocolitica, binds to the homodimeric secretion chaperone SycH (molecular surface rendering showing the distribution of electrostatic potential) in a manner that is very similar to the way in which the bacterial cytotoxin YopE (yellow ribbon) interacts with its cognate secretion chaperone SycE (not shown). SycH evidently unfolds YscM2 and presents it to the type III secretion apparatus in an extended conformation (p. 1591).
obituaries




Free 

research papers


DsbC forms a flexible dimer that can accommodate a wide variety of protein substrates for disulfide-bond isomerization.
PDB reference: DsbC, 1t3b, r1t3bsf

Dependence of ab initio connectivity-based phasing on its principal parameters allows an automated indication of the space group and of the number of molecules in the unit cell.
The crystal structure of apo protein tyrosine phosphatase 1B has been determined to 1.95 Å resolution. The structure shows four highly coordinated water molecules in the active site of the enzyme and the WPD-loop in a closed conformation.
PDB reference: apo protein tyrosine phosphatase 1B, 1sug, r1sugsf
Main-chain molecular fragments are positioned in electron density by a phased rotation and translation function. The fragments are refined as flexible generalized atoms and connected into chains. The sequence is aligned and side chains are built. The overall accuracy of the protein model is about 0.2 Å at a resolution of 1.2–1.9 Å.
To demonstrate a new strategy for engineering crystal lattices through metal-mediated interactions, apo acyl carrier protein has been crystallized in the presence of zinc ions. The structure was solved with MAD phasing using zinc anomalous signals.
PDB reference: apo acyl carrier protein, 1t8k, r1t8ksf
Shrimp alkaline phosphatase has been subjected to inhibitor and metal-exchange studies, which induced conformational changes in the active site.
A statistical design approach is proposed to screen for and optimize protein crystallization conditions.

The crystal structure of the first human parasite superoxide dismutase is described. Alternate positions for the catalytic copper are described along with differences within the electrostatic loops used for substrate guidance.

Linear interpolation of X-ray intensities from redundant data sets was used to determine the structures of insulin at different time points throughout the data collection. The structures reveal time-dependent structural changes that result from radiation exposure.
The crystal structure of the triple mutant (K53,56,120M) of a small protein (14 kDa), bovine pancreatic phospholipase A2, has been redetermined using sulfur single-wavelength anomalous scattering (S-SAS) at 1.6 Å resolution using a wavelength of 1.54 Å.
PDB reference: K53,56,120M PLA2, 1vkq, r1vkqsf

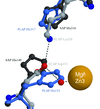
The structure of the type III secretion chaperone SycH from Y. pestis has been solved in complex with a stable fragment of YscM2, a negative regulator of type III secretion.
structural genomics papers
The structure of serine acetyltransferase from H. influenzae Rd determined at 2.7 Å resolution is presented. The structure is a hexamer consisting of a dimer of trimers, each of which contains subunits characterized by a conserved N-terminal α-helical domain and a C-terminal left-handed parallel β-helix.
PDB reference: serine acetyltransferase, 1s80, r1s80sf

The crystal structure of BAG-1 protein from C. elegans reveals a new BAG-domain fold.
PDB reference: BAG-1, 1t7s, r1t7ssf
crystallization papers

Transcription factor DksA from E. coli, which contains a canonical Zn-finger motif, was cloned expressed, purified and crystallized. The crystals diffract beyond 2.0 Å resolution at a synchrotron and three complete multiple anomalous dispersion data sets were collected at the absorption edge of the Zn atom.
The C-terminal domain CAPPD of the β-amyloid precursor protein, a type I transmembrane protein involved in Alzheimer's disease, was cloned, expressed in E. coli and, after purification, also crystallized. The initial electron-density map was of good quality and showed mostly helical regions.
The expression, purification, crystallization and preliminary X-ray diffraction studies of the gene product of cmcI from S. clavuligerus is reported. CmcI is believed to control the conversion of cephalosporins to cephamycins via 7α-methoxylation.
Carboxypeptidase Y inhibitor IC, complexed with the cognate proteinase, was crystallized and X-ray diffraction data were collected to 2.7 Å resolution.

Two new HIV proteases isolated from Brazilian patients failing intensive anti-AIDS therapy, namely subtype B mutant and subtype F wild type protein, were crystallized. The crystallization procedure and the molecular replacement solution for both proteins are presented in this paper.

Full-length SPN1 did not crystallize, so fragments with m3G cap binding activity were generated by protease cleavage. The crystals of a 26 kDa fragment which were obtained had to be enlarged by two rounds of microseeding.
Open access
access
The Sendai virus fusion-core six-helix bundle assembly was crystallized; crystals diffract X-rays to 2.5 Å resolution.

Crystallization and preliminary X-ray analyses of desulfurization enzyme DszB were conducted.

The main protein from the hemolymph of the cattle tick B. microplus has been crystallized using 1,6-hexanediol. The crystals belonged to the triclinic space group P1 and diffracted up to 2.1 Å resolution.

E. coli MutT was crystallized in binary and ternary complex forms. The crystals diffracted to 1.96 and 2.56 Å resolution, respectively.

The crystallization and preliminary X-ray analysis of heparinase II from P. heparinus is reported. Heparinases from P. heparinus are widely used in medical and analytical applications; however, their three-dimensional structures and detailed catalytic mechanisms have yet to be determined.
Triatoma virus (TrV) is a viral pathogen of blood-sucking reduviid bugs (triatomines). This work describes the purification of TrV particles from infected T. infestans and their crystallization and preliminary crystallographic analyses.

Human coactosin-like protein has been expressed in high yield and the His-tagged protein was purified by affinity chromatography. Several different crystal forms were obtained by the hanging-drop vapour-diffusion method.
Open access
access
The cofactor-binding domain of the LysR-type transcriptional regulator Cbl from E. coli has been cloned, overexpressed, purified and crystallized. The crystals diffract to 3.0 Å resolution using synchrotron radiation.
Open access
access
Three different crystal forms of apXPF were grown in trigonal, monoclinic and triclinic systems. The trigonal crystals diffracted to 2.8 Å and were grown in the presence of double-stranded DNA. Monoclinic crystals were grown without DNA and diffracted to 3.2 Å. Triclinic crystals were grown from a truncated apXPF protein lacking the tandem helix–hairpin–helix motifs and diffracted to 2.1 Å.
The N-terminal module of an archaebacterial threonyl-tRNA synthetase responsible for editing function has been crystallized in P31(2)21 space group.
Open access
access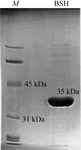
Two different crystal forms have been obtained of the conjugated bile salt hydrolase from B. longum and their preliminary crystallographic characterization has been carried out.

X29, a nuclear Nudix hydrolase that binds and decaps U8 snoRNA, has been expressed in high yield and crystallized in a form that diffracts to 2.1 Å resolution.
short communications

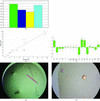
A solubility screen for determining the best solvent condition for a particular protein prior to crystallization trials is presented.
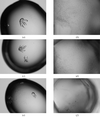
A solubility screen has been utilized to optimize initial crystallization results of A. pernix flap endonuclease-1.
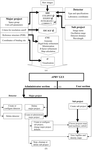
APRV is a program that automates routine procedures in high-throughput protein–ligand complex structure determination.
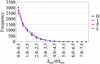
An algorithm for predicting the most informative wavelength combination in the MAD case is described.
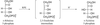
The X-ray crystal structure of D-ribulose 5-phosphate 3-epimerase (RPE) from Synechocystis reveals that the enzyme shares a highly conserved active site with other RPEs.
PDB reference: D-ribulose 5-phosphate 3-epimerase, 1tqj, r1tqjsf

CLIMS is a laboratory-information management system for protein crystallography that features a novel graphical interface to a relational database and assists all aspects of protein crystallization projects.
addenda and errata
Free

Addendum to the paper by Rao et al. (2004), Acta Cryst. D60, 1404–1412.