

 journal menu
journal menu





issue contents
October 2004 issue
Cover illustration: Pteridine reductase from parasitic trypanosomatid parasites is a potential drug target. A small inhibitor in the active site offers opportunities for modification to improve binding (p. 1780).
obituaries




Free 

research papers


Open access
access
The structure of the regulatory subunit of the protein kinase CK2 crystallized in the presence of p21WAF1 suggests binding in the solvent-accessible part of the zinc-finger motif. This new crystal form shows unreported conformations of the substrate-binding acidic loop.
PDB reference: CK2β–p21WAF, 1rqf, r1rqfsf
Methodologies are presented for systematizing and representing knowledge about the chemical and physical properties of additives used in crystallization experiments. A novel machine-learning and discovery program is introduced as a method that uses such knowledge for automatic analysis of augmented macromolecular crystallization data in order to categorize and find interesting relationships that can potentially aid the growth of new crystals.
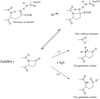
The high-resolution structure of succinyl-CoA:3-ketoacid CoA transferase in a new crystal form shows that the active-site glutamate residue adopts a conformation in which the enzyme would be susceptible to autolytic fragmentation when covalently linked to CoA. Succinyl-CoA:3-ketoacid transferase was crystallized in this new crystal form by changing the buffer to phosphate.
The structure of a lysozyme variant of 1309 non-H atoms that binds a single Rb+ ion (Z = 37) can be determined by direct methods using native data to 1.06 Å resolution. The results illustrate that proteins with more than 1000 atoms that include a halide-binding or other such site may be amenable to ab initio structure determination.
The three-dimensional structure of the complexes of S. griseus aminopeptidase (SGAP) with inhibitory L-tryptophan and p-iodo-L-phenylalanine have been determined and refined at 1.3 Å resolution. These structures confirm the previously proposed mode of action for SGAP and the critical enzyme residues involved in binding and catalysis.

The structure of reduced DsbC is analysed in terms of the factors that determine enzymatic activity.
PDB reference: DsbC, 1tjd, r1tjdsf

The crystal structure of PurE (N5-carboxyaminoimidazole ribonucleotide mutase) from the acidophile A. aceti compared with the structures of PurE from a known mesophile and thermophile allows the identification of important structural features that may confer acid-stability.
PDB reference: PurE, 1u11, r1u11sf

Three slightly different structures were determined by X-ray analysis of two crystal forms of the Fab from a humanized anti-γ-interferon antibody (HuZAF; USAN name fontolizumab), currently in phase II clinical trials for the treatment of Crohn's disease. The variable domain of the HuZAF heavy chain (VH) designed by a best-sequence-fit approach was structurally more similar to the murine precursor than a version (HuAF2) based on a human VH model with lower homology to the mouse sequence.
The structure of E. coli aminopeptidase P in complex with the inhibitor apstatin has been refined at 2.3 Å resolution. The structure of the complex illustrates how apstatin inhibits aminopeptidase P, but it does not offer an explanation for the proline-specificity of the enzyme.
PDB reference: aminopeptidase P–apstatin complex, 1n51, r1n51sf
Open access
access
A new crystal form of L. major pteridine reductase in complex with the inhibitor 2,4,6-triaminoquinazoline has been characterized. The inhibitor mimics the pterin head group of the archetypal antifolate methotrexate and despite being much smaller displays a similar inhibition constant. The structural detail may assist in the development of improved therapies for trypanosomal infections.
PDB reference: pteridine reductase ternary complex, 1w0c, r1w0csf
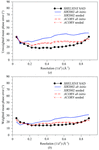
Electrostatic restraints as embodied by Coulombic models have generally proved deleterious to crystallographic refinement. Here, a Poisson–Boltzmann-based restraint is shown to be modestly beneficial to medium-resolution refinements.
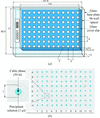
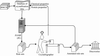
A relatively inexpensive laboratory-scale robotic system has been developed for the crystallization of membrane and soluble proteins in lipidic mesophases. It includes a robot for setting up crystallization trials, glass-based crystallization plates and an imaging robot. Under standard conditions, the robot can set up a 96-well plate in 13 min with just 20 nl protein solution per well.
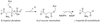
Mutant structures have been examined to establish the effect of perturbations in the active-site cysteine-histidine dyad on the catalytic efficiency of aspartate-β-semialdehyde dehydrogenase.
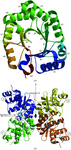
The crystal structure of 2-deoxyribose-5-phosphate aldolase from T. thermophilus HB8 has been solved at 1.5 Å resolution by the MAD method. The structure reveals a unique tetrameric oligomeric state, which is compared with those of two other aldolases from different organisms.
Crystals of the KatG C-terminal domain were produced in three different space groups, P212121, I41 and P1, depending on crystallization conditions and N-terminal sequence. The crystal packing in the P212121 space group presents a crystallographic novelty, with three pseudo-origin peaks reflecting a commensurate modulated packing.
Open access
access
Automated and accurate deposition of structures solved by X-ray diffraction to the Protein Data Bank
The RCSB Protein Data Bank (PDB) has a number of options for deposition of structural data and has developed software tools to facilitate the process. The options, procedures and tools for accurate and automated PDB data deposition are described here.
structural genomics papers
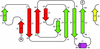
The structure of C. elegans SSP-19, an MSP-like domain, is reported. Sequence, structure and packing are compared to members of the MSP family.
PDB reference: SSP-19, 1row, r1rowsf
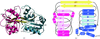
The crystal structure of a protein encoded by T. thermophilus HB8 gene TT1099 was solved to 1.75 Å using the multiple wavelength anomalous dispersion method and a selenomethionine incorporated protein, and the native protein structure was solved to 1.5 Å using a molecular replacement method. Both structures revealed a bound ligand, L-glutamate or L-glutamine, and a fold related to periplasmic substrate-binding proteins, indicating that the T. thermophilus HB8 molecule is most likely to be an L-glutamate or/and an L-glutamine-binding protein related to periplasmic receptors.

As part of a structural genomics project on bacterial gene products of unknown function, the crystal structures of YhdH, a putative quinone oxidoreductase, and its complex with NADP have been determined at 2.25 and 2.6 Å resolution, respectively.
crystallization papers

Farnesyl diphosphate synthase from T. brucei, the causative agent of African sleeping sickness, was expressed in E. coli and, after purification, crystallized in the absence and presence of the bisphosphonate inhibitor minodronate. The initial electron-density map was of good quality and showed mostly helical structure, but with the addition of two loop regions.

The purification, crystallization, data collection and preliminary structure solution of isoform α1 of the human thyroid hormone receptor LBD in complex with the thyroid hormones T3 and Triac in two crystal forms are reported.
Open access
access
Recombinant dipeptidyl peptidase IV from P. gingivalis has been crystallized. Data were collected to 2.7 Å from selenomethionine-derived crystals and structure solution by SAD or MAD is in progress.

Rv3291c, a putative Lrp/AsnC-family transcriptional regulator from M. tuberculosis, has been purified. The protein has been crystallized in the orthorhombic space group P2221 and the crystals diffract to 2.7 Å resolution.

Cellobiose phosphorylase from C. gilvus has been crystallized and X-ray diffraction data have been collected to 2.1 Å resolution at PF BL-5A.
Open access
access
Crystals of a fungal hydrolase with homology to penicillin-binding proteins have been grown by the sitting-drop vapour-diffusion technique from recombinant protein expressed in E. coli. Two crystal forms grow under different conditions. Diffraction data have been collected to 2.6 and 2.3 Å resolution.

This report describes the expression, purification, and crystallization of human Rheb, a small GTPase linking the tuberous sclerosis complex with the mTOR signaling pathway. Rheb has been crystallized in complexes with GDP, GTP, and a GTP analog GppNHp in multiple crystal forms which provide a basis for further structural and functional studies of Rheb.

YciE protein from E. coli was overexpressed and purified. Crystals of YciE belonged to space group R32 and diffracted to a resolution of 3.0 Å.

A putative agmatinase from D. radiodurans showing considerable sequence similarity to human agmatinase was crystallized. X-ray data have been collected to 1.80 Å.

The human phosphoglycerate mutase has been expressed in E. coli and crystallized by the hanging-drop vapor-diffusion method. The crystals belong to space group P21 and diffracted to a resolution of 2.8 Å.

The truncated mutant of EW29 from the annelid phylum earthworm L. terrestris comprising the C-terminal domain was crystallized. The crystal belonged to space group P43212, with unit-cell parameters a = b = 61.2, c = 175.6 Å, and diffracted to beyond 1.9 Å resolution.

A eukaryotic protein kinase inhibitor, CKI-7, has been cocrystallized with 3′-aminoglycoside phosphotransferase type IIIa. Crystals were obtained by microseeding.

The CP1 domain of Thermus thermophilus IleRS has been expressed in isolation, purified and cocrystallized with Val-AMS (a Val-AMP analogue) and with Val-2AA (a Val-tRNAIle analogue).
Open access
access
The class II hydrophobin HFBI has been purified and crystallized and X-ray diffraction data have been collected to 2.5 Å. This is the first crystallization report of the hydrophobin.

Vma5p (subunit C) from the V-ATPase of S. cerevisiae has been expressed, purified and crystallized. Diffraction data were collected to 1.80 Å from a native crystal and to 2.0 Å from a Lu-derivative crystal. In addition, rhombohedral crystals of the native protein were obtained that diffracted to 3.0 Å. The structure was solved using SIRAS.

The gcnA gene from S. gordonii, which confers N-acetyl-β-D-glucosaminidase activity, has been overexpressed. Crystals of GcnA diffract to 1.55 Å resolution and belong to space group P21212.
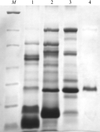
Crystals of the cysteine-rich secretory protein from N. atra are reported.

The leucyl-tRNA synthetase (LeuRS) from the archaeon P. horikoshii was overexpressed in a C-terminally truncated form in Escherichia coli, purified and crystallized by the hanging-drop vapour-diffusion method using ammonium sulfate as a precipitant.

Crystals of the outer membrane pyochelin receptor FptA from P. aeruginosa with pyochelin were obtained at high and low salt concentrations. They belong to space groups P1 and P212121, respectively. The orthorhombic crystals diffract beyond 2 Å resolution.

The crystallization and preliminary X-ray analysis of the lectin-like bacteriocin LlpA from Pseudomonas sp. BW11M1 is described. The crystals of recombinant protein expressed in E. coli belong to space group P21212, with unit-cell parameters a = 150.5, b = 154.5, c = 34.2 Å.

The P. abyssi homologue of Nip7p (PaNip7) was crystallized and X-ray data were collected from a native and an iodide-derivative crystal. Native crystals diffract to 1.8 Å and belong to space group C2, with unit-cell parameters a = 88.49, b = 90.28, c = 63.35 Å, β = 134.29°. The PaNip7 structure was solved using the SIRAS method.





