

 journal menu
journal menu





issue contents
July 2005 issue
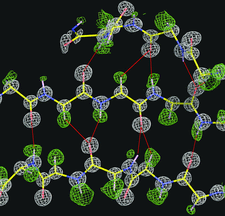
Cover illustration: The structure of air-oxidized P. abyssi rubredoxin mutant W4L/R5S solved by direct methods and refined by partially restrained full-matrix least-squares refinement to 0.69 Å resolution (p. 990).
obituaries




Free 

Obituary for M. Sundaralingam.
research papers
Open access
access
A robust method for determining bulk-solvent and anisotropic scaling parameters for macromolecular refinement is described. A maximum-likelihood target function for determination of flat bulk-solvent model parameters and overall anisotropic scale factor is also proposed.


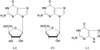
This work reports the structure of human PNP in complex with guanosine (at 2.80 Å resolution), 3′-deoxyguanosine (at 2.86 Å resolution) and 8-azaguanine (at 2.85 Å resolution). These structures were compared with the PNP–guanine, PNP–inosine and PNP–immucillin-H complexes solved previously.
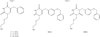
The structures of uridine phosphorylase complexed with several acyclic nucleoside analog inhibitors have been determined.

The method presented here evaluates protein crystallization states automatically.
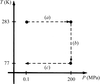
Protein crystals were successfully cryocooled by a He-gas high-pressure cooling method without any penetrative cryoprotectants. Excellent diffraction was observed. This method eliminates the need for penetrative cryoprotectants, thereby facilitating crystallographic data collection.

Small changes in temperatures during flash-cooling have significant effects on the diffraction quality of nucleosome crystals and in some cases cause substantial changes in unit-cell parameters that are a result of the reorganization of molecules in the lattice. Thus, in cases where flash-cooling around liquid-nitrogen temperatures leads to a deterioration in diffraction, flash-cooling at intermediate temperatures using alternative cryogens may be a useful approach.
Analysis shows that merging isomorphous replacement data with a limited number of highly accurate phases from the reference-beam diffraction experiment would significantly improve conventional experimental phasing.
The crystal structure of PEP carboxykinase from a succinate-producing bacterium has been solved in the native form and in complex with pyruvate, manganese and phosphate. The structure of the complex reveals 2-mercaptoethanol covalently bound to a cysteine residue located within a previously unreported conserved motif in the active-site cleft. This motif also contains an important interdomain connection.
Apo and cofactor-bound structures of T. thermophilus uroporphyrinogen-III C-methyltransferase solved to 2.0 Å resolution. These structures represent the `closed' form of the protein.
In vitro directed evolution of this enzyme produced mutants with enhanced activity towards a non-physiological substrate. X-ray crystallography was used to follow the resulting structural changes and explain the mechanistic implications.
PDB references: dienelactone hydrolase mutants, C123S, 1zi6; C123S, R206A, 1zic; E36D, 1zi9; E36D, C123S, A134S, S208G, A229V, K234R, 1zi8; E36D, R105H, C123S, G211D, K234N, 1zix; C123S bound with PMS, 1ziy; E36D, C123S bound with PMS, 1zj4; E36D, C123S, A134S, S208G, A229V, K234R bound with PMS, 1zj5

An incorrect HslV-HslU quaternary structure can result from two biologically functional complexes sitting at the interface of two coherently associated lattices.
PDB reference: HslV–HslU, 1yyf, r1yyfsf
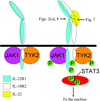
The crystal structure of glycosylated interleukin-22 has been determined at 2.6 Å resolution. The structure provides new insights into IL-22 receptor recognition.
PDB reference: insect-cell-expressed IL-22, 1ykb, r1ykbsf
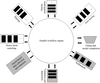
A method and a software suite based on it are described for searching both program and parameter space to find the best solution for a given X-ray diffraction data set of protein crystals.
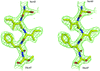
High-throughput de novo structure solution and refinement based on a single data set collected with chromium Kα radiation is described.
PDB reference: putative chorismate mutase, 1xho, r1xhosf

Structures of several proteins were solved from crystals that exhibited phenomena such as the simultaneous presence of two lattice types, pseudosymmetry accompanied by twinning and unusual types of non-isomorphism.

The statistical Shake-and-Bake procedure is optimized for Se-atom substructure determination.
Open access
access
The reflection profiles from glycerol kinase crystals were analyzed using highly monochromatic parallel synchrotron radiation and fine φ-slicing to elucidate the effects of flash-cooling on diffraction quality.

The structure of air-oxidized P. abyssi rubredoxin mutant W4L/R5S was solved by direct methods and refined by partially restrained full-matrix least-squares refinement to 0.69 Å resolution.
short communications
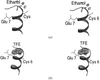
The effect of alcohols on protein structure has been studied by crystallography. Increasing solvent hydrophobicity is seen to be proportional to the overall dehydration of a protein's water shell.
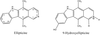
The 1.5 Å resolution crystallographic structure of a 6 bp DNA in complex with ellipticine confirms the intercalative binding previously described in a self-complementary ribodinucleoside monophosphate and enables an accurate measurement of the distortion effects of the drug on real DNA.
PDB reference: ellipticine–DNA complex, 1z3f, r1z3fsf





