journal menu
journal menu





issue contents
September 2005 issue
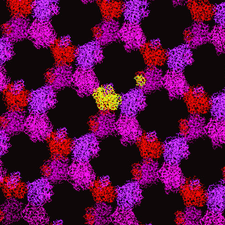
Cover illustration: Packing of the tetragonal crystal form of the EETI-II-![[beta]](/logos/entities/beta_rmgif.gif) TNNK complex (p. 1255).
TNNK complex (p. 1255).
topical reviews




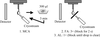
This review provides a compilation of post-crystallization methods that can convert poorly diffracting crystals into data-quality crystals. Protocols for annealing, dehydration, soaking and cross-linking are outlined and examples of some spectacular changes in crystal quality are provided.
research papers


The structure of the periodate complex of lysozyme has been determined, refined and analyzed.
PDB reference: periodate complex of lysozyme, 1hc0, r1hc0sf

The crystal structures of oxidised and reduced nitrite reductase from R. sphaeroides give new insight into the pH dependence of nitrite binding to the active site.
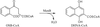
The crystal structures of naphthoate synthase (MenB) and its complex with the product naphthoyl-CoA have been determined by X-ray crystallography at resolutions of 2.15 and 2.30 Å, respectively. The native structure shows several flexible regions around the active site which are only partly rigidified by the bound ligand.

The crystal structure of a putative 2′-5′ RNA ligase from P. horikoshii was solved and compared with a Thermus thermophilus homologue and the structurally related cyclic phosphodiesterase.
PDB reference: putative 2′-5′ RNA ligase, 1vdx, r1vdxsf
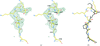
The structure of P. falciparum GAPDH has been solved to 2.6 Å resolution with one molecule of NAD+ bound per subunit. Amino-acid substitutions which cluster in a groove about the R dyad suggest a potential binding site for haem, which is known to specifically inhibit the Plasmodium enzyme.
PDB reference: glyceraldehyde-3-phosphate dehydrogenase, 1ywg, r1ywgsf
Open  access
access
access
The X-ray structure of the enzyme 5-aminolaevulinic acid dehydratase (ALAD) from yeast complexed with the competitive inhibitor 5-hydroxylaevulinic acid has been determined at a resolution of 1.9 Å.
PDB reference: 5-aminolaevulinic acid dehydratase, 1w31, r1w31sf
Open access
access
A simple method for treating RIP data is presented which improves the quality of RIP substructures as well as the phases from downstream phasing applications.
Open access
access
The problems encountered during the phasing and structure determination of the packaging enzyme P4 from bacteriophage φ13 which arise from the arrangement of the four hexamers comprising the crystallographic asymmetric unit are described.
Open access
access
The malarial purine nucleoside phosphorylase (PNP) in complexes with inosine and with sulfate, are reported. These structures are compared with a previous structure of P. falciparum PNP in complex with a transition-state inhibitor.
Open access
access
The cystine-knot protein EETI-II is shown to be particularly rigid.
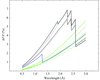
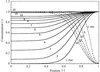
Based on the evaluation of a large number of experiments carried out at different X-ray wavelengths with respect to the anomalous signal-to-noise ratio, the optimum wavelength for diffraction data collection was found to be close to 2.1 Å irrespective of the nature of the anomalously scattering substructure.
The structure of the antibiotic resistance factor AAC(6′)-Ii in complex with coenzyme A has been determined in a new crystal form. Comparison of this structure with the previously determined complex and in combination with normal-mode analysis reveals conformational plasticity that may be important to enzymatic mechanism.
PDB reference: AAC(6′)-Ii, 2a4n, r2a4nsf
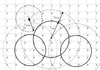
The use of hydrogel beads for the crystallization of proteins is explored in this contribution.

Analysis of data collected from Hg-derivatized crystals reveals that mercury–cysteine bonds are very susceptible to radiation damage.
short communications
It is shown that the standard bulk-solvent corrrection, as implemented in CNS, can lead to incorrect results in the case of large macromolecular structures. A modified method of the solvent mask definition is proposed.
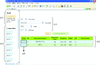
Heavy-atom Database System is a program that assists heavy-atom derivatization of proteins. On the basis of amino-acid sequence/motif and crystallization conditions, the program suggests potential heavy-atom reagents for any target protein by searching against a database of known heavy-atom derivatives.

Surface-entropy reduction was used to obtain high-quality crystals of human choline acetyltransferase.
addenda and errata
Free

Open access
access