journal menu
journal menu





issue contents
January 2006 issue
Data collection and analysis
Proceedings of the CCP4 study weekend
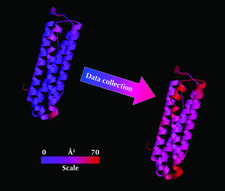
Cover illustration: B-value increase inflicted by X-ray irradiation of a cryocooled crystal of apoferritin between sequential data sets 1 and 10 at beamline ID14-4, ESRF, Grenoble. Structures are coloured from blue (0 Å2) to red (70 Å2) (see p. 32).




research papers
Open  access
access
access
The present situation and possible future developments of macromolecular crystallography are reviewed.
Open access
access
This article aims to give synchrotron users an overview of the functioning of a synchrotron beamline and how the performance of various instruments combines to allow the collection of diffraction data.
Open access
access
This article reviews the progress in the field of optimization of genetic constructs for improved soluble protein expression and provides a general overview of relevant mutation methods, screens and selections.
Open access
access
A review discussing some of the techniques that may extend the diffraction limits or otherwise improve the quality of the X-ray diffraction data from a crystal.
Open access
access
Macromolecular cryocrystallographic methods and their rationale are reviewed, and our current limited understanding of radiation damage in cryocooled crystals is summarized.
Open access
access
Processing a set of diffraction images can be divided into three steps: autoindexing, accurate cell determination and finally integration. The basic procedures involved in each of these steps are described.
Open access
access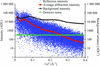
Quantitative evaluation of the dependency of data-set statistics on the data-collection parameters are presented.
Open access
access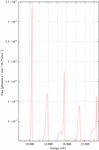
Guidelines for the collection of macromolecular crystallographic data at undulator X-ray sources are discussed.
Open access
access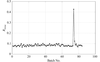
Putting intensity data on a consistent scale and analysing the agreement to determine data quality and determination of Laue group symmetry from observed intensities are described.
Open access
access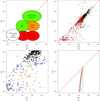
Analysis of the Protein Data Bank February 2004 release using such simple statistics as the R factor between potentially twin-related reflections identified cases with twinning. Careful consideration of these showed that noncrystallographic symmetry and twinning often occur together, causing serious problems with the determination of true crystal symmetry.
Open access
access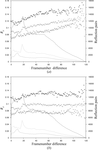
Aspects of analysis and correction of radiation damage in macromolecular crystallography.
Open access
access
Automated methods of protein crystallization, data collection and crystallographic computing have had a significant impact on the throughput of protein–ligand structures generated to support drug-discovery process. The high turnover of cocrystallization and crystal-soaking experiments often results in partially damaged crystals producing imperfect and difficult to process diffraction patterns, which, after careful processing, can reveal valuable structural information.


Open access
access
The methodology used in the structure determination of human semaphorin 4D based on MAD phasing is discussed.
PDB reference: semaphorin 4D, 1olz, r1olzsf
Open access
access
Entropic effects play a critical role in protein crystallization. Mutational engineering of the surface of proteins aimed at the reduction of excess surface conformational entropy has potential to become an effective method to enhance the crystallizability of many proteins.