journal menu
journal menu





issue contents
June 2006 issue
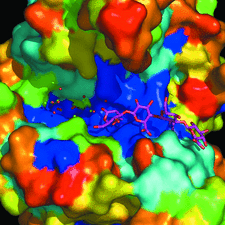
Cover illustration: The figure illustrates conservation of amino-acid residues in the active site of hyaluronidases from glycoside hydrolase family 56 (p. 595). The surface is coloured by sequence entropy (dark blue means fully conserved while red means not conserved).
feature articles




Free 

This article reviews the issues, methods and strategies involved in obtaining the crystals of protein-ligand complexes that are critical for structure-aided drug design.
research papers


Open access
access
A high-resolution structure determination shows that (+)-epi-biotin and (+)-biotin bind similarly to streptavidin by using conformational changes in their tetrahydrothiophene rings to accommodate their different configurations at the C2 carbon.
The crystal structure of human dual-specificity protein tyrosine phosphatase 18 provides the structural basis for the unusual catalytic activity at high temperature.
PDB reference: DSP18, 2esb, r2esbsf

The structures of the PDI-related protein Wind and three mutants show the same mode of dimerization and only subtle differences in the putative Pipe-binding region, although two of the mutants completely abrogate Pipe transport.
Open access
access
This paper describes the production and structural characterization of the allergen Ves v 2 from wasp venom. Implications for cross-reactivity are discussed.
PDB reference: Ves v 2, 2atm, r2atmsf
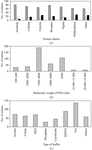
A survey of crystallization conditions was carried out for all known protein–protein complexes to define the boundaries of their configuration space. Based on the survey, a probability-based sparse-matrix screen and a systematic buffer and pH screen are proposed to facilitate the crystallization of protein–protein complexes.
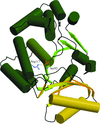
Various binary complex structures of E. coli malonyl-CoA–acyl carrier protein transacylase are presented, including that of the natural substrate malonyl-CoA. Based on the presented structural data, a possible new catalytic enzyme mechanism is discussed.
The structure of Rom has been determined in a new crystal form from X-ray diffraction data to 2.5 Å resolution.
PDB reference: ColE1 Rom protein, 2ghy, r2ghysf

The structure of a complex of M. tuberculosis pantothenate kinase with a coenzyme A derivative has been determined in two crystal forms, each at two temperatures. The structure and a comparative analysis involving the known structure of the E. coli enzyme and the sequences of the enzyme from other bacteria show a strong correlation among structural plasticity, evolutionary relatedness and function.
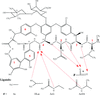
Intermolecular interaction energies between fragments of glycopeptide antibiotics and small peptide ligands are evaluated using X-ray geometries and new methods, suitable for application to very large molecular complexes. The calculation of the electrostatic contributions is based on charge densities constructed with a databank of transferable aspherical atoms, whereas non-electrostatic contributions are evaluated with a new set of atom–atom potentials fitted to symmetry-adapted perturbation-theory results.
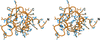
Upon introduction of an X-ray pseudo-energy derived from the electron density present at a specific protein residue, protein design algorithms can be used readily for crystallographic model building and refinement purposes.
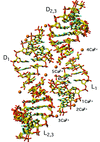
The racemate of the RNA duplex r(CUGGGCGG)·r(CCGCCUGG) crystallizes with L-duplexes and D-duplexes stacked end-to-end in a centrosymmetric lattice.
PDB references: RNA racemate, 2g32, r2g32sf; 2gq4, r2gq4sf; 2gpm, r2gpmsf; 2gq5, r2gq5sf; 2gq6, r2gq6sf; 2gq7, r2gq7sf
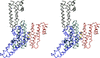
The structure of the plant invertase inhibitor Nt-CIF from tobacco has been determined in six independent crystal forms covering pH 4.6–9.5.

The high-resolution structure of a new crystal form of human cytoglobin has been determined, revealing an additional helix in the N-terminus and a new dimeric arrangement.
PDB reference: human cytoglobin, 2dc3, r2dc3sf
Open access
access
Revisions are based on structures now in the Protein Data Bank with resolution ≤1.25 Å, as well as small-molecule structures in the Cambridge Structural Database; patterns shown in bidentate carboxylates are explored.
short communications

The crystal structure of LeACX1 from tomato was determined to 2.74 Å resolution. The packing arrangement observed was unusual, containing three monomers in the asymmetric unit: the biological dimer plus a half dimer.
PDB reference: LeACX1, 2fon, r2fonsf