journal menu
journal menu




issue contents
July 2006 issue
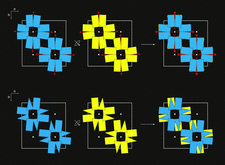
Cover illustration: The figure illustrates how the crystal packing of Rna1p enabled hemihedral twinning (p. 750). Twinned crystal growth was possible owing to the presence of a twofold non-crystallographic symmetry axis almost parallel to the twin operator.
research papers




A method has been developed for readily preparing crystals for noble-gas SAD phasing and cryopreservation and has been demonstrated with the 240-residue 26 kDa protein porcine pancreas elastase (PPE). The diffraction quality of the method is sufficiently high that a single 0.31 occupied krypton site was sufficient for SAD phasing at 1.3 Å resolution.


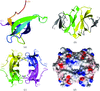
The first high-resolution structure (1.1 Å resolution) of a trimethoprim-resistant plasmid-encoded R67 DHFR is described. Sheets of pentagonal networks of water molecules in the D2-symmetric active site may play a role in ligand binding, catalysis and inhibition.
PDB reference: dihydrofolate reductase, 2gqv, r2gqvsf
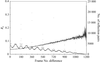
Sulfur SAD data analyses reveal that successful substructure solution and phasing is a sensitive function of choices made regarding the length of data collection and resolution cutoffs.
The paper reports two forms of the crystal structure of a quadruple mutant (K53,56,120,121M) of PLA2. The two structures of the quadruple mutant, monoclinic and trigonal, represent the conformations of PLA2 at the lipid interface and in solution, respectively.

The crystal structure of the human Rab protein Rab6B in its inactive (with bound MgGDP) and active (MgGTPγS-bound) forms have been determined to 2.3 and 1.8 Å, respectively. Both crystallized in space group P212121, with similar unit-cell parameters.
The structure of the E. coli EntA protein, a bacterial member of the short-chain oxidoreductase family of enzymes, has been solved at 2.0 Å. Analysis of core residues from other family members allows the identification of active-site residues.
PDB reference: EntA, 2fwm, r2fwmsf
Open  access
access
access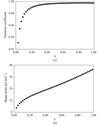
An automated computational procedure for fitting a ligand into its electron density with the use of the MMFF94 force field and a Gaussian shape description has been developed.

This case study shows that partial merohedral twinning does not hinder MIR phasing if it is recognized and the phasing and refinement procedures are adjusted accordingly. Identification of hemihedral twinning, twin correction and different refinement strategies for twinned data are discussed.
PDB reference: Rna1p, 2ca6, r2ca6sf

Flash-freezing and/or ligand binding can cause changes not only in crystal packing but also in the size and shape of the internal cavities in the protein molecule.

Download citation


Download citation
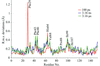
X-ray crystal structures of transient intermediate states after the photolysis of carbon monoxide from myoglobin reveal a non-linear progression toward the deoxy state.
PDB references: myoglobin, crystal 1, `laser off', 2g0r, r2g0rsf; crystal 2, `laser off', 2g0s, r2g0ssf; 100 ps coordinate model, 2g0v, r2g0vsf; 316 ps coordinate model, 2g0x, r2g0xsf; 1 ns coordinate model, 2g0z, r2g0zsf; 3.16 ns coordinate model, 2g10, r2g10sf; 31.6 ns coordinate model, 2g11, r2g11sf; 316 ns coordinate model, 2g12, r2g12sf; 3.16 µs coordinate model, 2g14, r2g14sf
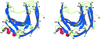
The crystal structure of an orthorhombic form of xylanase II was determined at atomic resolution and its structural fluctuation was evaluated by analyzing the anisotropic displacement parameters.

The first crystal structure of a C2 domain of rabphilin-3A is reported.
PDB reference: rabphilin-3A C2A domain, 2chd, r2chdsf
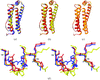
High-resolution X-ray structures of human ferritin L chain in two different crystal forms revealed new insights in the mechanism of iron storage.
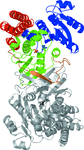
The crystal structure of M. tuberculosis 1-deoxy-D-xylulose 5-phosphate reductoisomerase, with a sulfate bound in the expected site of the phosphate moiety of the sugar substrate, has been determined to 1.9 Å resolution. Structure and sequence comparisons offer suggestions for the design of specific drugs.

The structure of the apo form of the enzyme with sulfate ions bound in the active site is presented. The structure is in a `closed' conformation relative to structures of the P. vulgaris enzyme.
PDB reference: E. coli tryptophanase, 2c44, r2c44sf

The crystal structure of 8Sα globulin of mung bean was determined at 2.65 Å resolution and its physicochemical properties were elucidated in comparison with other seed storage globulins.
PDB reference: 8Sα globulin, 2cv6, r2cv6sf

A preliminary screen has been used to maximize solubility and define buffer and salt conditions for optimal protein crystal growth of ten test proteins. A precipitant/precipitant–additive screen is also presented which is used in conjunction with the preliminary screen to define a two-step approach to crystal screening.