journal menu
journal menu





issue contents
November 2006 issue
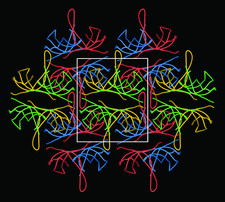
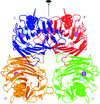
Cover illustration: Packing of molecules in the crystal structure of Thermus aquaticus single-stranded DNA-binding protein (p. 1407). The N-terminal domains are in blue and green and the C-terminal domains are in red and yellow. The blue/red molecules form one layer in the xz plane, related by the twofold axis parallel to the crystal y axis (of C2221 symmetry); the green/yellow molecules form another layer, related by the 21 axis with respect to the previous layer.
research papers






Open  access
access
access
The crystal structure of internalin C from L. monocytogenes is reported. The putative receptor-binding surface is flat and predominantly hydrophilic and the Ig-like domain has exposed aromatics that may be of functional importance.
PDB reference: internalin C, 1xeu, r1zeusf
Open access
access
The crystal structure of the hydrolase domain of human 10-formyltetrahydrofolate dehydrogenase, an important enzyme in the metabolism of one-carbon units, has been determined. The active site was further expored by solving the structure of a complex containing a substrate fragment.
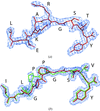
Human MHC-I molecules are currently subdivided into twelve different supertypes, each presenting a distinct part of the sequence space of peptides for immune recognition. Here, the first structures of peptide–MHC complexes involving a member of the B62 HLA supertype are presented.

A new crystallization database has been created, which can be analyzed to develop crystallization strategies.

The crystal structures of the complexes of basic winged-bean lectin with galactose, 2-methoxygalactose, N-acetylgalactosamine and methyl-α-N-acetylgalactosamine provide a structural rationale for the carbohydrate-specificity of the lectin and insights into the differential affinity exhibited by some plant lectins for galactose and N-acetylgalactosamine.
A database of invarioms for structural refinement of amino-acid, oligopeptide and protein molecules is presented.

The mail-in crystallography program at the NSLS has been very successful for several years. With the advent of our new database (PXDB), the operation and the advantages brought to the PXRR group is now revisited from the users' point of view.

This manuscript chronicles the evolution of software used originally to control a diffractometer at a macromolecular crystallography beamline.
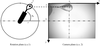
C3D is a program for automated crystal centring with improved performance in crystal-centre detection using visible light.

Software for automated crystal centring on beamlines for macromolecular crystallography is described.
The crystal structure of reindeer β-lactoglobulin is reported. Structure solution and refinement were significantly complicated by almost perfect pseudo-body-centering.
PDB reference: reindeer β-lactoglobulin, 1yup, r1yupsf

The crystal structure of B. pertussis BugE solute receptor is reported at a resolution of 2.3 Å. BugE has a bound fortuitous ligand, identified as a glutamate. Ligand binding in the Bug family of proteins is discussed.
PDB reference: BugE, 2dvz, r2dvzsf
This first report of a homodimeric E1 apoenzyme structure shows no evidence for disorder-to-order loop transformations relative to the holoenzyme, in contrast to what has been observed in the heterotetrameric branched-chain-specific enzyme. In the active-site region, the histidine residue His142 clearly exhibits partial occupancy of two distinct rotamer sites in the apoenzyme and the number of ordered solvent molecules increases from apoenyme to holoenzyme to inhibitor complex.
PDB reference: pyruvate dehydrogenase E1 component, 2g67, r2g67sf
Two crystal structures of endonuclease I from V. cholerae identify a chloride buried in the core of the enzyme. Magnesium, which is essential for catalysis, is bound to the enzyme at neutral pH but not at low pH.

Structures of tetracycline complexed with trypsin-modified EF-Tu are reported.
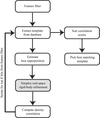
Incorporating a Nelder–Mead Simplex search into the side-chain identification and model-building routines of TEXTAL.

The crystal structure of the single-stranded DNA-binding protein from T. aquaticus was solved and refined at 1.85 Å resolution.
PDB reference: SSB, 2fxq, r2fxqsf
Crystal structures of the complexes of peanut lectin with Galβ1-3Gal, methyl-T-antigen, Galβ1-6GalNAc, Galα1-3Gal and Galα1-6Glc and that of a crystal obtained in the presence of Galα1-3Galβ1-4Gal have been determined.
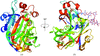
The structure of laminarinase Lam16A from the wood-degrading fungus P. chrysosporium was solved to 1.34 Å resolution. It is the first structure in the non-specific 1,3(4)-β-D-glucanase subfamily of glycoside hydrolase family GH16.
PDB reference: laminarinase Lam16A, 2cl2, r2cl2sf
short communications
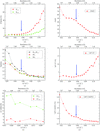
The crystal structure of VEGF-E has been solved by S-SAD using highly redundant data. To obtain the highest data accuracy, special care was taken in setting up the beam parameters for data collection.
PDB reference: VEGF-E, 2gnn, r2gnnsf