

 journal menu
journal menu





issue contents
January 2007 issue
Crystallography of complexes
Proceedings of the CCP4 study weekend
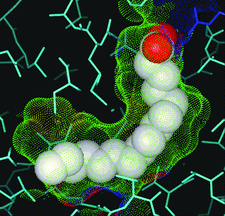
Cover illustration: Nuclear receptor 1 with bound natural ligand (p. 72).




research papers
Open  access
access
access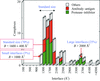
X-ray structures in the PDB illustrate both the specific recognition of two polypeptide chains in protein–protein complexes and dimeric proteins and their nonspecific interaction at crystal contacts.
Open access
access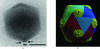
The most extensive structural information on viruses relates to apparently icosahedral virions and is based on X-ray crystallography and on cryo-electron microscopy single-particle reconstructions. This paper concerns itself with the study of the macromolecular complexes that constitute viruses, using structural hybrid techniques.
Open access
access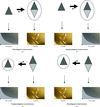
Structures of protein complexes offer some of the most interesting insights into biological processes. In this article, the methods required to show that the complex observed is the physiological one are investigated.
Open access
access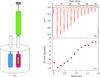
The use of isothermal titration calorimetry (ITC) provides a full thermodynamic characterization of an interaction in one experiment. The determination of the affinity is an important value; however, the additional layer of information provided by the change in enthalpy and entropy can help in understanding the biology. This is demonstrated with respect to tyrosine kinase-mediated signal transduction.
Open access
access
Four case studies in using maximum-likelihood molecular replacement, as implemented in the program Phaser, to solve structures of protein complexes are described.
Open access
access
A method for detecting structural homologs of components in an intermediate resolution cryo-EM map and their spatial configuration is presented.
Open access
access
This paper presents a survey of techniques that explore the surface properties of protein:protein interfaces so as to inform the prediction of probable sites of protein:protein interaction on newly determined protein structures.
Open access
access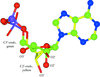
A brief summary of the types of restraint defined in refinement dictionaries.
Open access
access
This paper highlights some of the problems that can arise when attempting to obtain crystal structures of small molecule–protein complexes and how biophysical methods can be used to define and overcome these problems. Many of the techniques mentioned are also applicable to the study of protein–protein complexes and mode-of-action analysis.
Open access
access
Methods presented for growing protein–ligand complexes fall into the categories of co-expression of the protein with the ligands of interest, use of the ligands during protein purification, cocrystallization and soaking the ligands into existing crystals.


Open access
access
A case study showing how the determination of multiple cocrystal structures of the protein tyrosine kinase c-Abl was used to support drug discovery, resulting in a compound effective in the treatment of chronic myelogenous leukaemia.
Open access
access
Methods and resources for obtaining chemically plausible starting models and restraint sets for refinement of ligand complexes are described and some of the potential pitfalls are discussed.
Open access
access
An automated ligand-fitting procedure is applied to (Fo − Fc)exp(iφc) difference density for 200 commonly found ligands from macromolecular structures in the Protein Data Bank to identify ligands from density maps.
Open access
access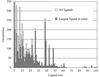
The performance of the ligand-building module of the ARP/wARP software suite is assessed through a large-scale test on known protein–ligand complexes. The results provide a detailed benchmark and guidelines for future improvements.







