

 journal menu
journal menu




issue contents
March 2007 issue
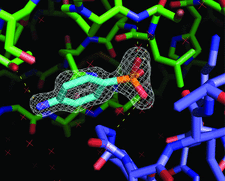
Cover illustration: A sulfanilic acid molecule creates a lattice interface in a crystal of porcine trypsin (p. 310).
editorial



Free 

research papers


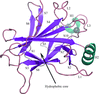
The crystal structure of the human p53 core domain in the absence of DNA has been determined with a new monoclinic space group (P21). Detailed analysis of non-crystallographic symmetry and crystal packing reveals that the p53 molecules are arranged differently from those in other p53 crystals, suggesting the possible existence of different p53 core domain tetramers.
PDB reference: human p53 core domain, 2ocj, r2ocjsf
The structure of a CooA variant from the thermophilic bacterium Carboxydothermus hydrogenoformans (ChCooA) is reported in which one monomer is fully in the on-state.
PDB reference: CooA, 2hkx, r2hkxsf

The structures of S. cerevisae glutaredoxin 1 and of a fusion protein consisting of yellow fluorescent protein and glutaredoxin were solved by molecular replacement and refined to 2.0 and 2.7 Å, respectively.
Crystal structures with and without His-tags have been surveyed and the authors show that His-tags are not hazardous to your structure.
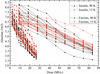
A small but significant reduction of X-ray-induced radiation damage at 15 K compared with 90 K could be found in the systematic study of a total of 54 crystals of insulin and holoferritin.
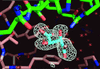
Difference Fourier X-ray diffraction analyses of nine crystals are presented, which convincingly demonstrate the validity of the hypothesis that protein crystallization can be driven by the inclusion of small molecules rich in hydrogen-bonding, hydrophobic and electrostatic bonding possibilities.
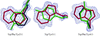
Structure of double-stranded right-handed double-helical gramicidin channels (dimers) in the crystal of gramicidin D with KI complex has been determined. High-resolution data enabled quantitative determination of gramicidin components and distributions of the I anions outside and K cations inside the channel, which was confirmed by their anomalous scattering.
PDB reference: gramicidin D with KI, 2izq, r2izqsf
Open access
access
The crystal structures of T. gondii and P. falciparum ENR in complex with NAD+ and triclosan and of T. gondii ENR in an apo form have been solved to 2.6, 2.2 and 2.8 Å, respectively.

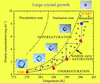
A methodology and an instrument for the temperature-controlled optimization of crystal growth are described. The technique finds application in the growth of large high-quality crystals for neutron crystallography.
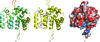
The SAD technique was used to determine the crystal structure of a putative redox-enzyme maturation protein from A. fulgidus at 3.4 Å resolution.
PDB reference: AF0173, 2o9x, r2o9xsf

The crystal structure of glyoxylate reductase from the hyperthermophilic archaeon P. horikoshii OT3 complexed with NADP(H) was solved at 1.7 Å resolution. The present study revealed its cofactor-recognition and thermostability mechanisms.
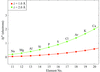
The anomalously scattering substructures in 23 crystals of 19 different biological macromolecules have been determined based on diffraction data collected at a wavelength of 2.0 Å.
PDB references: anomalous substructures of apoferritin, 2g4h, r2g4hsf; concanavalin A, 2g4i, r2g4isf; glucose isomerase, 2g4j, r2g4jsf; human ADP-ribosylhydrolase 3, 2g4k, r2g4ksf; lysozyme, pH 4.5, 2g4p, r2g4psf; pH 8.0, 2g4q, r2g4qsf; hydroxynitrile lyase, 2g4l, r2g4lsf; insulin, 2g4m, r2g4msf; α-lactalbumin, 2g4n, r2g4nsf; 3-isopropylmalate dehydrogenase, 2g4o, r2g4osf; MogA, 2g4p, r2g4psf; NBR1 PB1, 2g4s, r2g4ssf; porcine pancreatic elastase, Na form, 2g4t, r2g4tsf; Ca form, 2g4u, r2g4usf; proteinase K, 2g4v, 2g4vsf; ribonuclease A, C2, 2g4w, r2g4wsf; P3221, 2g4x, r2g4xsf; thaumatin, 2g4y, r2g4ysf; thermolysin, 2g4z, r2g4zsf; titin-(A168-A169), 2ill, r2illsf; trypsin, P1, 2g51, r2g51sf; P21, 2g52, r2g52sf; P3121, 2g54, r2g55sf
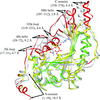
The structure of a transition-state analog of muscle creatine kinase reveals significant asymmetry within the functional homodimer. The amino-terminal region is shown to be intimately involved in subunit association and intersubunit communication.
PDB reference: muscle creatine kinase, 1u6r, r1u6rsf

The crystal structure of the AlaX-M trans-editing enzyme revealed that the conserved glycine-rich loop in the N-terminal domain is located near the catalytic site in the C-terminal domain.
PDB reference: AlaX-M, 2e1b, r2e1bsf

Download citation


Download citation
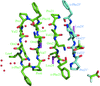
Gramicidin S, a membrane-active antibiotic, was crystallized from solvent containing water, methanol, trifluoroacetic acid and hydrochloric acid. The structure was refined at 0.95 Å resolution and contains 1.5 molecules of gramicidin S, two trifluoracetic acid molecules and ten water molecules in the crystallographic asymmetric unit. In the crystals, the gramicidin S molecules line up into helical channels that differ from those observed previously.
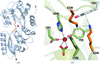
The crystal structures of two mutants of the N-terminal half-molecule of human transferrin, K206E and K206E/K296E, have identified important features of the mechanism of iron release.
short communications

Structures of human galectin-3 carbohydrate-recognition domain determined at various stages of ligand removal provided insight into ligand-diffusion rates and their significance with respect to ligand-exchange protocols.
books received
Free

letters to the editor
Free

international union of crystallography
Free








