

 journal menu
journal menu





issue contents
January 2008 issue
Molecular replacement
Proceedings of the CCP4 study weekend
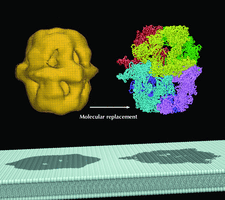
Cover illustration: Using an EM map as a model for molecular replacement. Left: an EM map of yeast fatty acid synthase (FAS) at 24 Å resolution. Right: an X-ray crystal structure of FAS determined at 4 Å resolution. Bottom: a schematic representation of the lipid bilayer whose major component is the fatty acid synthesized by FAS (p. 76).




research papers
Open  access
access
access
An outline is given of the basic features of the molecular-replacement method for solving crystal structures.
Open access
access
An account is given of the latest developments of the AMoRe package.
Open access
access
This review outlines questions to consider when attempting to solve crystal structures by molecular replacement.
Open access
access
The problems of gaining accurate protein sequence alignments for molecular replacement are discussed, current techniques explained and strategies suggested.
Open access
access
The default model-preparation scheme of MOLREP is described. Two examples are presented of model improvement using X-ray data.
Open access
access
The possibility of taking into account large-amplitude collective movements of a given model by using a subset of low-frequency normal modes is evaluated for molecular replacement and refinement using X-ray data or cryo-EM maps.
Open access
access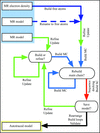
A systematic test shows how ARP/wARP deals with automated model building for structures that have been solved by molecular replacement. A description of protocols in the flex-wARP control system and studies of two specific cases are also presented.
Open access
access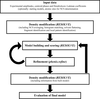
The highly automated PHENIX AutoBuild wizard is described. The procedure can be applied equally well to phases derived from isomorphous/anomalous and molecular-replacement methods.
Open access
access
Overview and examples of combined use of X-ray and electron-microscopy data.
Open access
access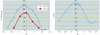
Test studies have been conducted on five crystal structures of large molecular assemblies, in which EM maps are used as models for structure solution by molecular replacement using various standard MR packages such as AMoRe, MOLREP and Phaser.
Open access
access
A number of techniques for the location of small and medium-sized model fragments in experimentally phased electron-density maps are explored. The application of one of these techniques to automated model building is discussed.
Open access
access
Three difficult MR cases are reported in which the orientation of a search oligomer or its internal parameters were determined and the oligomer was positioned according to the maximal value of the correlation coefficient in a series of translation searches. Such an exhaustive search was feasible because of constraints on the model parameters derived from the self-rotation function.

Open access
access
The presence of pseudosymmetry can cause problems in structure determination and refinement. The relevant background and representative examples are presented.
Open access
access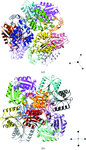
The type II dehydroquinase enzyme is a symmetrical dodecameric protein which crystallizes in either high-symmetry cubic space groups or low-symmetry crystal systems with multiple copies in the asymmetric unit. Both systems have provided challenging examples for molecular replacement; for example, a triclinic crystal form has 16 dodecamers (192 monomers) in the unit cell. Three difficult examples are discussed and two are used as test cases to compare the performance of four commonly used molecular-replacement packages.
Open access
access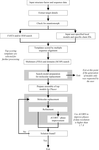
An automation pipeline for macromolecular structure solution by molecular replacement with a special emphasis on the discovery and preparation of a large number of search models is described.
Open access
access
The fully automated pipeline, BALBES, integrates a redesigned hierarchical database of protein structures with their domains and multimeric organization, and solves molecular-replacement problems using only input X-ray and sequence data.
Open access
access
The practical limits of molecular replacement can be extended by using several specifically designed protein models based on fold-recognition methods and by exhaustive searches performed in a parallelized pipeline. Updated results from the JCSG MR pipeline, which to date has solved 33 molecular-replacement structures with less than 35% sequence identity to the closest homologue of known structure, are presented.







