journal menu
journal menu





issue contents
June 2009 issue
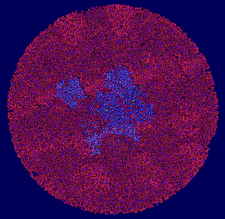
Cover illustration: The presence of translational pseudosymmetry, a large number of molecules in the asymmetric unit (eight) and a poor search model complicated the structure solution of Desulfovibrio desulfuricans (ATCC 29577) flavodoxin (p. 523). Structure solution in a second crystal form, P43, provided an improved model that along with complimentary approaches to molecular replacement facilitated determination of an initial solution. Phaser located six copies in the asymmetric unit, but generation of a 100 Å sphere of crystallographic symmetry-related molecules showed large spaces that corresponded with the presence of unoccupied density in a 2Fo - Fc composite OMIT map; together these analyses suggested the presence of additional molecules in the asymmetric unit. Two additional copies were placed manually using clear density for the FMN molecule that had been excluded from the search model and the pseudo-translational symmetry operators as guides. A 100 Å sphere of crystallographic symmetry-related molecules (pink) generated after manual placement of the final two molecules within the asymmetric unit (light blue) to analyze the final packing is shown.
research papers






 access
access

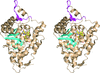 access
access
 access
access
 access
access access
access
short communications