journal menu
journal menu





issue contents
July 2009 issue
Includes papers presented at the workshop on New Algorithms in Macromolecular Crystallography and Electron Microscopy, Leiden, The Netherlands
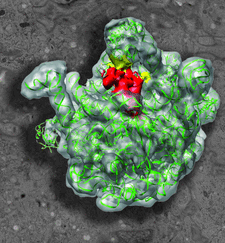
Cover illustration: Heat shock protein 15 bound to 50S ribosome superimposed on an electron micrograph.
new algorithms workshop




Open  access
access
access
A summary of the 2008 New algorithms in macromolecular crystallography and electron microscopy Max-Inf2/Lorentz Center workshop in Leiden is given.
Open access
access
An algorithm is described that calculates the most likely primitive unit cell given a set of randomly oriented electron-diffraction patterns with unknown angular relationships.
Open access
access
The application of a new normal-mode-based X-ray crystallographic refinement method to a total of eight structures of moderate resolution is illustrated.
Open access
access
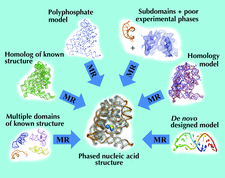
Molecular replacement with the simultaneous use of several search functions may solve the phase problem when the conventional molecular-replacement procedure fails to identify the solution.
Open access
access
UROX is software designed for the interactive fitting of atomic models into electron-microscopy reconstructions. The main features of the software are presented, along with a few examples.
Open access
access
An introduction to the current paradigm shift towards concurrency in software.
Open access
access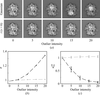
An expectation-maximization algorithm for maximum-likelihood refinement of electron-microscopy data is presented that is based on finite mixtures of multivariate t-distributions. Compared with the conventionally employed Gaussian mixture model, the t-distribution provides robustness against outliers in the data.
Open access
access
This paper describes procedures for obtaining confidence intervals for coordinate locations resulting from the fitting of atomic models into low-resolution densities.
Open access
access
The interpretation of a 20 Å resolution electron-density map using segmentation and pattern-recognition-based identification of domain shapes is described.
research papers
The room-temperature X-ray structures of two proteins, solved at 1.8 and 1.9 Å resolution, are used to investigate whether a set of conformations, rather than a single X-ray structure, provides better agreement with both the X-ray data and the observed 13Cα chemical shifts in solution.


In order to gain a detailed understanding of the structure–function relationship of T. cruzi dihydrofolate reductase (DHFR), the three-dimensional structure of this protein in complex with various ligands is being studied. Here, the crystal structures of T. cruzi DHFR-TS with three different compositions of the DHFR domain are reported: the folate-free state, the complex with the lipophilic antifolate trimetrexate and the complex with the classical antifolate methotrexate.
The crystal structures of three isoforms of anionic trypsin from chum salmon reveal a structural basis for their differences in catalytic efficiency and explain how small differences in sequence and structure allow enzymes to perform their biological functions in a wide range of environments.
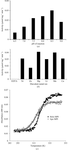
The X-ray crystal structures of mannose-6-phosphate isomerase from S. typhimurium in the apo form (with no metal bound), the holo form (with zinc), bound to yttrium (at an inhibitory site) and complexed with the cyclic form of the substrate fructose 6-phosphate and Zn2+ are reported.