journal menu
journal menu





issue contents
August 2009 issue
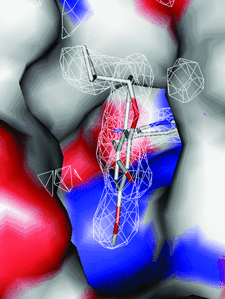
Cover illustration: Inhibitor binding to the TMP-resistant F98Y single-mutant dihydrofolate reductase from S. aureus (p. 751). The final 2Fo - Fc difference electron density contoured at 1.5 Å is shown as white mesh and is superimposed with the structure of AR-101 as observed in a ternary complex with NADPH. The molecular surface of the active site is indicated and coloured by electrostatic potential: blue, positive; red, negative. Unoccupied density corresponds to water molecules.
research papers







The structure of R75A-mutant rat parvalbumin solved by molecular replacement points out the role of the Arg75–Glu81 salt bridge in attaching the nonbinding AB domain to the rest of the protein core.
PDB reference: R75A rat parvalbumin, 3f45, r3f45sf
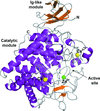
Here, the crystal structure of an endoglucanase, Cel9A, from Alicyclobacillus acidocaldarius (Aa_Cel9A) is reported which displays a modular architecture composed of an N-terminal Ig-like domain connected to the catalytic domain. This paper describes the overall structure and the detailed contacts between the two modules.
PDB reference: Aa_Cel9A, 3ez8, 3ez8sf
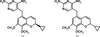
The binding modes of DHFR inhibitors AR-101, AR-102 and iclaprim have been analysed and compared. The X-ray crystallographic data explain the binding modes of all molecules well and can be used to rationalize the equipotent affinity of AR-101, AR-102 and iclaprim, which is also reflected in their antibacterial properties.
Open  access
access
access
The structural determination procedures of a gap junction channel at 3.5 Å resolution are described, including the preparation of crystals, intensity data collection, data processing, phasing and structural refinement.
PDB reference: gap junction channel, 2zw3, r2zw3sf

The 1.9 Å resolution crystal structure of TTHA1846 from T. thermophilus HB8 complexed with coenzyme A and zinc ion is reported. The putative function of TTHA1846 and the inhibitory role played by the zinc ion in its activity are discussed.
PDB reference: TTHA1846, 2cye, r2cyesf

The structure of the C119S,C162S double mutant of human MAP-kinase p38β has been solved and refined to 2.05 Å resolution. A comparison of the structures of p38α and p38β shows that they are highly similar overall but that there are small conformational differences that could be exploited in order to achieve drug selectivity.
Open access
access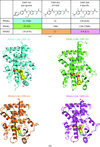
The structures of the ligand-binding domains (LBDs) of human peroxisome proliferator-activated receptors (PPARα, PPARγ and PPARδ) in complexes with a pan agonist, an α/δ dual agonist and a PPARδ-specific agonist were determined. The results explain how each ligand is recognized by the PPAR LBDs at an atomic level.
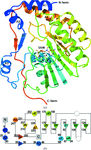
The structure of Modoc virus NS5 methyltransferase reveals structural conservation between methyltransferases in different flaviviral evolutionary groups. These findings support the choice of the Modoc virus as a model for flaviviral studies.
The crystal structure of A. aeolicus cytochrome c555 (AA c555) has been determined at 1.15 Å resolution. A unique helix structure is revealed which might be responsible for the high stability of AA c555.
PDB reference: AA c555, 2zxy, r2zxysf
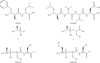
The structure of an APN–aminophosphinic inhibitor complex is reported.
PDB reference: APN–PL250 complex, 2zxg, r2zxgsf
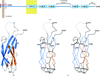
In SAD phasing, the efficacy of cocrystallization versus soaking in the binding of the different Gd complexes, their mode of interaction and a comparative study using synchrotron radiation and rotating-anode generator, were tested.
The crystal structure of the complex between ristocetin A and the cell-wall peptide mimetic N-acetyl-lysine-D-alanine-D-alanine has been solved. Structural details explaining the anticooperativity of the antibiotic have been identified.
Open access
access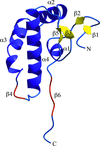
The structure of the cytosolic C-terminal domain of nonstructural protein 4 from feline coronavirus has been determined and analyzed.
PDB reference: C-terminal domain of nsp4, 3gzf, r3gzfsf

In order to probe the role of residue 96, three P. falciparum triosephosphate isomerase mutants, F96S, F96H and F96W, were biochemically and structurally characterized. The studies show that Phe96 and Ser73 are important for maintaining the optimal hydration of the active site required for ligand binding. A new binding site for the ligand at the dimer interface has also been identified.
The crystal structure of the Calx-β domain of integrin α6β4 at 1.48 Å resolution provides the first example of a non-calcium-binding Calx-β domain.
short communications

The crystal structure of PDE10a in complex with papaverine was generated by ligand soaking of protein crystals that were cross-linked by glutaraldehyde.
PDB reference: PDE10a–papaverine complex, 2wey, r2weysf

The structure of the HipBA complex from E. coli has been solved using the SAD technique applied to a mercury derivative. A novel HipA activation mechanism is proposed and an intriguing tryptophan-binding pocket on the surface of the protein is described.
PDB reference: HipBA, mercury derivative, 2wiu, r2wiusf