journal menu
journal menu





issue contents
July 2010 issue
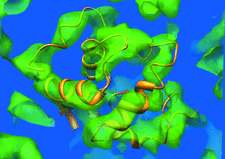
Cover illustration: Electron-density map (green) for hen egg-white lysozyme resulting from a multiple isomorphous replacement analysis applied to X-ray powder diffraction data, superimposed onto the known molecular structure (orange) (p. 756).
research papers






The process of structure determination of the minimal complex between Tfb2 and Tfb5, which are subunits of the transcription/DNA-repair factor TFIIH, is described, illustrating how domain boundaries can influence crystal packing and thus crystal quality and diffraction.
Molecular structural information on hen egg-white lysozyme was obtained up to approximately 5.3 Å resolution by applying the de novo method of multiple isomorphous replacement to powder diffraction data. At this resolution, features of the secondary structure of lysozyme can be located in the electron-density map representing the protein molecule of correct chirality.

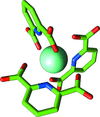
The binding mode of a luminescent lanthanide complex used for solving protein structures by anomalous diffraction methods is fully described.
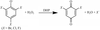
X-ray crystal structures of the metcyano form of dehaloperoxidase-hemoglobin (DHP A) from Amphitrite ornata (DHPCN) and the C73S mutant of DHP A (C73SCN) have been determined in order to further investigate the geometry of diatomic ligands coordinated to the heme iron.
A multivariate likelihood function for phase combination that accounts for the correlation between experimental and density-modified electron-density maps leads to the automatic building of many more crystal structures compared with current methods.
The effectiveness of salts of organic acids as potential cryoprotective agents is presented.

The high-resolution crystal structure of porcine succinyl-CoA:3-oxoacid coenzyme A transferase (SCOT) shows a glycerol molecule hydrogen bonded to the catalytic glutamate of one subunit of one of the two dimers in the asymmetric unit. This subunit also shows dynamic domain movements which may be mechanistically relevant.
PDB reference: succinyl-CoA:3-oxoacid-coenzyme A transferase, 3k6m

The crystal structure of the dimethyllysine derivative of the E. coli RNA polymerase α subunit C-terminal domain is reported at 2.0 Å resolution.
PDB reference: RNA polymerase α subunit C-terminal domain, 3k4g

It was clarified biochemically that only one of the two homologues of glutamyl-tRNA synthetase (GluRS) functions as a nondiscriminating (ND) GluRS in Thermotoga maritima. Furthermore, the crystal structure of the T. maritima ND–GluRS in complex with a Glu-AMP analog was determined at 2.0 Å resolution.
PDB reference: nondiscriminating glutamyl-tRNA synthetase, 3afh

Two apo crystal structures and a GTP-bound crystal structure of MoaC protein from T. thermophilus HB8 have been determined at 1.9, 1.75 and 2.5 Å resolution, respectively. In addition, isothermal titration calorimetry experiments and molecular-dynamics simulations have been carried out in order to study the protein–ligand interactions.
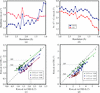
A stereochemical library which defines the target values for main-chain bond lengths and angles as a function of the residue's φ/ψ angles was tested in refinement. Use of this library allows the construction of models that conform to ideal geometry much better than previous libraries without degrading their fit to the diffraction data.
Crystal structures of ribonuclease U2 at 0.96 and 0.99 Å resolution imply that the isomerization of Asp45 to isoaspartate is promoted by the water molecules bound to the surface of the protein.
short communications
The structure of cytochrome c552 reveals that its flexibility is important for its function.
PDB reference: cytochrome c552, 3m97