journal menu
journal menu





issue contents
December 2010 issue
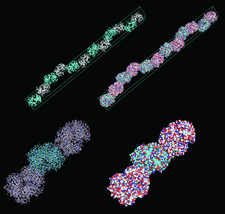
Cover illustration: Structure of a crystal form of human methaemoglobin (p. 1316). The contents of the hexagonal unit cell and the filament that it defines are shown in ribbon and surface representations, along with an outline of the unit cell for reference. Details consisting of three haemoglobin molecules and the two kinds of interfaces that relate them are also shown. The filaments display 6122 symmetry with a periodicity of 677.5 Å.
editorial




Free 

The problem of undercounting of citations that are published only in supplementary material is studied for the journals Nature, Science, Cell and the Proceedings of the National Academy of Sciences (USA).
research papers


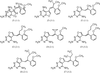
The structures of six chirally mixed E/Z-isomers of 5-substituted 2,4-diaminofuro[2,3-d]pyrimidines reveals only one isomer is bound in the active site of human DHFR. The configuration of all but one C9-analogue is observed as the E-isomer.
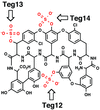
The 2.7 Å resolution crystal structure of Teg14, a glycopeptide sulfotransferase cloned from an uncultured soil bacterium, is described. The relationship of Teg14 to other sulfotransferases is discussed.
PDB reference: Teg14, 3nib
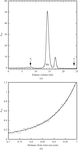
Measurements of the penetration depth and distribution of photoelectron damage excited by 18.6 keV photons in a lysozyme crystal are reported for the first time.

The crystal structure of the autotransporter Hbp missing domain 2 is presented. The role of domain 2 is unknown, but domain 1 is shown to bind calcium ions.
PDB reference: Hbp, 3ak5
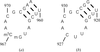
The crystal structure of PH0851, an archaeal homologue of the bacterial Fmu/RsmB/RrmB rRNA cytosine 5-methyltransferase, has been determined.
PDB reference: PH0851, 2yxl
Crystallographic and biochemical analyses of the recombinant bacterial lipocalin, Blc, provide evidence for its native state as a functional monomer, as for most other natural lipocalins. The previously reported dimeric structure of a modified version of Blc (Blc-X) seems to be a result of an N-terminal extension arising from the applied cloning strategy.
PDB reference: monomeric Blc, 3mbt

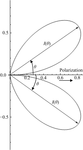
A novel form of crystalline human methemoglobin has been solved that has 12 molecules of the protein comprising helical turns of about 675 Å pitch. The possible relationship of this helix to sickle cell hemoglobin fibrils is discussed.
PDB reference: methemoglobin, 3odq
The crystal structure of actinidin from A. arguta Planch. (sarunashi) was determined by the SAD method using in-house Cu Kα and Cr Kα radiation.
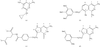
High-resolution crystal structures of T. brucei pteridine reductase in complex with four ligands are described. The ligands include the known antifolates pemetrexed and trimetrexate, a 2,4-diaminopyrimidine derivative and cyromazine, a melamine derivative of veterinary importance. The structures provide accurate information about new molecular entities in the active site of a potential drug target against protozoan parasites.
Open access
access
The angle τ (backbone N—Cα—C) is the most contested Engh and Huber refinement target parameter. It is shown that this parameter is `correct' as a PDB-wide average, but can be improved by taking into account residue types, secondary structures and many other aspects of our knowledge of the biophysical relations between residue type and protein structure.

The 1.9 Å resolution structure of FOXO4-DBD–DNA complex is reported and compared with other known structures of FOXO-DBD–DNA complexes.
PDB reference: FOXO4-DBD–DNA complex, 3l2c