journal menu
journal menu





issue contents
July 2012 issue
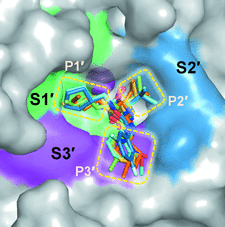
Cover illustration: Surface diagram of Staphylococcus aureus peptide deformylase (PDF) with the active-site pocket depicted with four inhibitors (p. 784). The molecular surface is coloured green (S1'), blue (S2') and magenta (S3'). The P1', P2' and P3' residues of the four inhibitors corresponding to the S1', S2' and S3' sites are indicated by yellow dashes. The Zn2+ ion is shown as a sphere
research papers







The structure of a xylanase from F. oxysporum (FoXyn10a) was determined at 1.94 Å resolution. FoXyn10a adopts the (β/α)8-barrel fold of the GH10 family, with an extended loop above the catalytic site with potential implications for enzymatic function.
PDB reference: FoXyn10a, 3u7b

The crystal structure of the complex between the nuclear transport factor importin-α and the peptide corresponding to the nuclear localization sequence from flap 1 endonuclease (FEN1) has been determined. The structure rectifies the binding mode previously proposed and therefore explains the functional results that were previously controversial.
PDB reference: importin α–FEN1NLS complex, 3uvu
Succinyl-CoA synthetase catalyzes the reaction succinyl-CoA + NDP + Pi ⇌ succinate + CoA + NTP, where N denotes adenosine or guanosine. The enzyme from T. aquaticus was characterized biochemically and its structure was determined in complex with GDP-Mn2+, the preferred nucleotide.
PDB reference: succinyl-CoA synthetase, 3ufx
X-ray structures of S. solfataricus serine:pyruvate aminotransferase in different intermediate states and in complex with inhibitor have increased our understanding of the enzyme mechanism and have allowed an insight into the substrate specificity of this industrially important enzyme.
Two crystal structures of UMPK from Helicobacter pylori in complex with GTP and with UDP are reported.
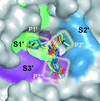
Crystal structures of peptide deformylase from S. aureus in complex with novel inhibitors are presented together with their functional analysis. A detailed comparative analysis of these structures as well as the activities of the inhibitors allowed the elucidation of distinctive structural changes which depend upon the class of inhibitor.
The structure of potato serine protease inhibitor, the most abundant protease inhibitor in potatoes, is reported and sheds light on the inhibition mechanism of this protein.
PDB reference: PSPI, 3tc2

A bond-distance analysis to determine the protonation states of ionizable amino acids has been made for trypsin at 1.2 Å resolution, subtilisin at 1.26 Å resolution and lysozyme at 0.65 Å resolution. This was effective for Asp and Glu but not for His.
Open  access
access
access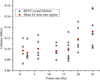
A systematic increase in lifetime is observed in room-temperature protein and virus crystals through the use of reduced exposure times and a fast detector.

Crystal structures of the family 3b carbohydrate-binding module (CBM3b) of the cellulosomal multimodular hydrolytic enzyme cellobiohydrolase 9A (Cbh9A) from C. thermocellum and of its mutant Cbh9A CBM3bN126W have been determined at 2.20 and 1.04 Å resolution, respectively. This mutation reforms the cellulose-binding properties of the adhesion-deficient CBM.

The single mutation E193D of the catalytic acid/base of Neotermes koshunensis β-glucosidase makes it favour the transglycosylation reaction. The NkBgl E193D mutant may be suitable for development as a useful tool for the enzymatic synthesis of numerous therapeutic oligosaccharides.
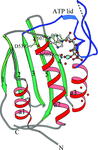
The histidine WalK (YycG) plays a crucial role in coordinating murein synthesis with cell division and the crystal structure of its ATP binding domain has been determined. Interestingly the bound ATP was not hydrolyzed during crystallization and remains intact in the crystal lattice.
PDB reference: WalK451–ATP complex, 3sl2

The crystal structure of a putative general stress protein from the citrus canker bacterium X. citri pv. citri was determined to 2.5 Å resolution.
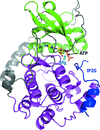
ATP bound in the active site of protein kinase A is readily hydrolysed to ADP and free phosphate by X-ray irradiation at room temperature. The phosphate ion observed in the active site causes a dramatic conformational change of the bound peptide inhibitor.
Open access
access
A density-based procedure is described for improving a homology model that is locally accurate but differs globally. The model is deformed to match the map and refined, yielding an improved starting point for density modification and further model-building.
addenda and errata
Free

book reviews
Free