journal menu
journal menu





issue contents
May 2014 issue
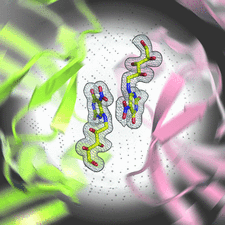
Cover illustration: The active site of the enzyme riboflavin synthase from the pathogenic bacterium Brucella abortus, with two molecules of the product analogue 5-nitro-6-ribitylamino-2,4(1H,3H)-pyrimidinedione bound in an antiparallel way (p. 1419). Interestingly, the active site of this enzyme is formed by two substrate-binding sites bearing pseudo-C2 symmetry that are located at the interface between two neighbouring chains.
research papers






Open  access
access
access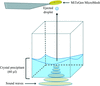
A method is presented for screening fragment libraries using acoustic droplet ejection to co-crystallize proteins and chemicals directly on micromeshes with as little as 2.5 nl of each component. This method was used to identify previously unreported fragments that bind to lysozyme, thermolysin, and trypsin.
Open access
access
Two crystal structures of the major pilin SpaD from C. diphtheriae have been determined at 1.87 and 2.5 Å resolution. The N-terminal domain is found to contain an isopeptide bond that forms slowly over time in the recombinant protein. Given its structural context, this provides insight into the relationship between internal isopeptide-bond formation and pilus assembly.

An improved integration method enhances the quality of electron-density maps and decreases the number of diffraction patterns that are needed in serial femtosecond crystallography.

Crystal structures show how bacterial PTPS homologues possess an unusual activity that differs from that of mammalian PTPS enzymes.

Open access
access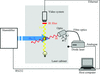
A novel method and the associated instrumentation for improving crystalline order (higher resolution of X-ray diffraction and reduced mosaicity) of protein crystals by precisely controlled heating is demonstrated. Crystal transformation is optically controlled by a video system.
Open access
access
Semiempirical quantum-chemical X-ray macromolecular refinement using the program DivCon integrated with PHENIX is described.
Open access
access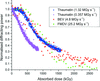
A departure from a linear or an exponential decay in the diffracting power of macromolecular crystals is observed and accounted for through consideration of a multi-state sequential model.

The first comprehensive analysis of the sunitinib malate crystal structure and charge-density distribution in the context of sunitinib complexes with a series of protein kinases is reported. The aspherical atom databank approach, Hirshfeld surface analysis and quantum theory of atoms in molecules are used as a foundation for the investigations of interactions between an inhibitor and a protein.
The structure of a medium-chain dehydrogenase reductase-type glucose dehydrogenase showing narrow substrate specificity was determined, and the unique structural features of the enzyme were compared with those of a glucose dehydrogenase showing broad substrate specificity.
The crystal structure of T. acidophilum IdeR is reported.
High-resolution X-ray crystallography has been effectively combined with resonance Raman spectroscopy to `fingerprint' and validate different redox and ligand states in structures of Alcaligenes xylosoxidans cytochrome c′.
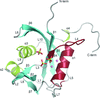
The structures of Escherichia coli YmfB on its own and in complex with three sulfates and two manganese ions, when compared with the structures of other Nudix hydrolases such as MutT, Ap4Aase and DR1025, provide insight into the unique hydrolysis mechanism of YmfB.
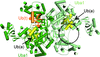
The structure of a ternary complex consisting of the ubiquitin-activating enzyme Uba1 and two differently bound ubiquitin molecules has been determined by X-ray crystallography. For the first time, the formation of the ubiquitin-acyladenylate could be structurally visualized together with the attachment of a second ubiquitin covalently linked to the active-site cysteine of Uba1. Model-building studies provide insights into the recruitment of ubiquitin-conjugating enzymes.
Open access
access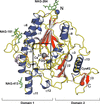
Structure of sulfamidase provides insight into the molecular pathology of mucopolysaccharidosis IIIA
Mucopolysaccharidosis IIIA is a fatal neurodegenerative disease that typically manifests itself in childhood and is caused by mutations in the gene for the lysosomal enzyme sulfamidase. The first structure of this enzyme is presented, which provides insight into the molecular basis of disease-causing mutations, and the enzymatic mechanism is proposed.
A single mutation in the linker region renders intact P22 tailspike crystallizable, allowing analysis of the crystal structure of the complete tailspike at high resolution. The mutant protein is still functional, but has to be distorted to fit into a published cryo-EM map, and the structure suggests an important role of the highly conserved linker in intramolecular signal transmission.
Open access
access
Flexible torsion angle-based NCS restraints have been implemented in phenix.refine, allowing improved model refinement at all resolutions. Rotamer correction and rotamer consistency checks between NCS-related amino-acid side chains further improve the final model quality.
Open access
access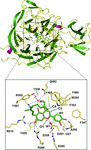
The crystal structure of sialidase from C. perfringens, a pathogenic bacterium causing various gastrointestinal diseases, was determined in complex with a potent natural polyphenolic geranylated flavonoid-based inhibitor. The complex structure and comparative kinetic studies revealed that the geranyl group and C3′ hydroxyl group of the flavonoid backbone contribute to inhibition of the bacterial sialidase and generation of the stable enzyme–inhibitor complex.
PDB reference: Cp-NanI catalytic domain, complex with diplacone, 4l2e

The first structure of a CAZy glycoside hydrolase family 52 enzyme, that of G. thermoglucosidasius β-xylosidase, reveals structural similarities to other families with which it shares less than 13% sequence identity. The substrate-bound complex confirms the active-site residues and is consistent with a retaining mechanism.
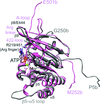
The crystal structure of KaiC from Thermosynechococcus elongatus allows insight into the different anatomies of the CI and CII ATPases.
PDB reference: KaiC, 4o0m
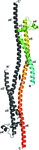
CagL mediates contact of the pathogen H. pylori with host-cell integrins. A domain-swapped structure of CagL shows the RGD motif to act as a hinge region embedded in a helix and reveals the structure of the functionally important C-terminus.
PDB reference: CagL, 4cii
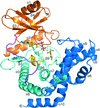
The seven crystal structures presented in this work represent the first multiple reaction complexes (GlcNAc/GalNAc, GlcNAc-1P/GalNAc-1P, ATP/ADP and Mg2+) of NahK in the lacto-N-biose (LNB)/galacto-N-biose (GNB) pathway, providing the molecular basis for the recognition of substrates and gaining insights into the catalytic mechanism at the molecular level.
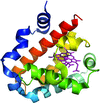
The first crystal structure of a class 3 nonsymbiotic plant haemoglobin is reported to 1.77 Å resolution. The structure revealed a novel N-terminal helical extension that participates extensively in the dimeric interface.
PDB reference: AHb3, 4c0n
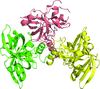
This work reports crystal structures of trimeric riboflavin synthase from the pathogen B. abortus both as the apo protein and in complex with several ligands of interest. It is shown that ligand binding drives the assembly of the unique active site of the trimer, and these findings are complemented by a detailed kinetic study on this enzyme, in which marked inhibition by substrate and product was observed.
Open access
access
The crystal structure of a previously unsolved type of cypovirus polyhedrin has been determined from data collected directly from frozen live insect cells.
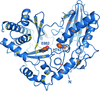
The synthesis of nonribosomal peptides often introduces D-configured amino acids into the final products. The first crystal structure of the epimerization domain of a nonribosomal peptide synthetase reveals a bilobal architecture and implies a general acid–base mechanism for the L↔D isomerization of aminoacyl substrates.
PDB reference: E domain of TycA, 2xhg
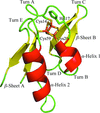
The crystal structure of the ferredoxin HaPuxC has been solved at 2.3 Å resolution.
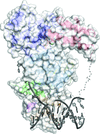
The crystal structure of the human BLM helicase in complex with ADP and a 3′-overhang DNA duplex is reported.
PDB reference: BLM, complex with ADP and duplex DNA, 4o3m
Open access
access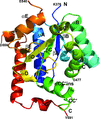
Crystal structure of the SEFIR domain from human IL-17 receptor A provides new insights into IL-17 signaling.
PDB reference: IL-17 receptor A SEFIR domain, 4nux
Open access
access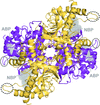
A crystal structure of the human lactate dehydrogenase A apo form suitable for ligand soaking and a NADH binary-complex structure are presented.
short communications
Here, new evidence is provided to show that the inclusion of weak-intensity, high-resolution X-ray diffraction data helps to improve the quality of experimental phases by imposing proper constraints on electron-density models during noncrystallographic symmetry averaging.
PDB reference: YfbU, 4lr3
addenda and errata
Free