The title compound, pentabarium tris(vanadate) chloride, crystallizes in the well known apatite structure type, with space group
P6
3/
m and
Z = 2. The crystal structure contains isolated (VO
4)
3- tetrahedra that are bridged by Ba
2+ ions. The intermediate Cl
- anions, situated on positions with
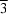
symmetry, are octahedrally surrounded by Ba
2+ cations.
Supporting information
Key indicators
- Single-crystal X-ray study
- T = 298 K
- Mean
![[sigma]](//journals.iucr.org/logos/entities/sigma_rmgif.gif) (V-O) = 0.004 Å
(V-O) = 0.004 Å
- R factor = 0.015
- wR factor = 0.034
- Data-to-parameter ratio = 14.2
checkCIF/PLATON results
No syntax errors found
 Alert level B
GOODF01_ALERT_2_B The least squares goodness of fit parameter lies
outside the range 0.60 <> 4.00
Goodness of fit given = 0.461
Alert level B
GOODF01_ALERT_2_B The least squares goodness of fit parameter lies
outside the range 0.60 <> 4.00
Goodness of fit given = 0.461
 Alert level C
ABSTM02_ALERT_3_C The ratio of expected to reported Tmax/Tmin(RR) is > 1.10
Tmin and Tmax reported: 0.170 0.220
Tmin and Tmax expected: 0.130 0.225
RR = 1.334
Please check that your absorption correction is appropriate.
PLAT042_ALERT_1_C Calc. and Rep. MoietyFormula Strings Differ .... ?
PLAT060_ALERT_3_C Ratio Tmax/Tmin (Exp-to-Rep) (too) Large ....... 1.30
PLAT086_ALERT_2_C Unsatisfactory S Value (Too Low or Not Given) .. 0.46
PLAT141_ALERT_4_C su on a - Axis Small or Missing (x 100000) ..... 10 Ang.
Alert level C
ABSTM02_ALERT_3_C The ratio of expected to reported Tmax/Tmin(RR) is > 1.10
Tmin and Tmax reported: 0.170 0.220
Tmin and Tmax expected: 0.130 0.225
RR = 1.334
Please check that your absorption correction is appropriate.
PLAT042_ALERT_1_C Calc. and Rep. MoietyFormula Strings Differ .... ?
PLAT060_ALERT_3_C Ratio Tmax/Tmin (Exp-to-Rep) (too) Large ....... 1.30
PLAT086_ALERT_2_C Unsatisfactory S Value (Too Low or Not Given) .. 0.46
PLAT141_ALERT_4_C su on a - Axis Small or Missing (x 100000) ..... 10 Ang.
 Alert level G
FORMU01_ALERT_1_G There is a discrepancy between the atom counts in the
_chemical_formula_sum and _chemical_formula_moiety. This is
usually due to the moiety formula being in the wrong format.
Atom count from _chemical_formula_sum: Ba5 Cl1 O12 V3
Atom count from _chemical_formula_moiety:
Alert level G
FORMU01_ALERT_1_G There is a discrepancy between the atom counts in the
_chemical_formula_sum and _chemical_formula_moiety. This is
usually due to the moiety formula being in the wrong format.
Atom count from _chemical_formula_sum: Ba5 Cl1 O12 V3
Atom count from _chemical_formula_moiety:
0 ALERT level A = In general: serious problem
1 ALERT level B = Potentially serious problem
5 ALERT level C = Check and explain
1 ALERT level G = General alerts; check
2 ALERT type 1 CIF construction/syntax error, inconsistent or missing data
2 ALERT type 2 Indicator that the structure model may be wrong or deficient
2 ALERT type 3 Indicator that the structure quality may be low
1 ALERT type 4 Improvement, methodology, query or suggestion
Data collection: SMART (Bruker, 2002); cell refinement: TOPAS (Bruker, 2002); data reduction: SAINT (Bruker, 2002); program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure: CRYSTALS (Betteridge et al., 2003); molecular graphics: ATOMS (Dowty, 2000); software used to prepare material for publication: CRYSTALS.
Pentabarium tris(vanadate) chloride
top
Crystal data top
| Ba5(VO4)3Cl | Dx = 4.732 Mg m−3 |
| Mr = 1066.92 | Mo Kα radiation, λ = 0.71073 Å |
| Hexagonal, P63/m | Cell parameters from 526 reflections |
| Hall symbol: -P 6c | θ = 7.5–70° |
| a = 10.5565 (1) Å | µ = 14.94 mm−1 |
| c = 7.7584 (1) Å | T = 298 K |
| V = 748.76 (1) Å3 | Polyhedron, colourless |
| Z = 2 | 0.15 × 0.12 × 0.10 mm |
| F(000) = 924 | |
Data collection top
Bruker SMART APEX-2
diffractometer | 567 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.04 |
| ω scans | θmax = 28.2°, θmin = 3.4° |
Absorption correction: multi-scan
(SADABS; Bruker, 2002) | h = −13→10 |
| Tmin = 0.17, Tmax = 0.22 | k = −13→9 |
| 3252 measured reflections | l = −10→5 |
| 661 independent reflections | |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Method of quasi-unit weights, w = [1/(2*Fobs)]2 |
| R[F2 > 2σ(F2)] = 0.015 | (Δ/σ)max = 0.001 |
| wR(F2) = 0.034 | Δρmax = 0.68 e Å−3 |
| S = 0.46 | Δρmin = −0.52 e Å−3 |
| 567 reflections | Extinction correction: Larson (1970) |
| 40 parameters | Extinction coefficient: 76.8 (16) |
| 0 restraints | |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Ba1 | 0.3333 | 0.6667 | −0.00077 (6) | 0.0099 | |
| Ba2 | 0.25686 (4) | 0.01113 (4) | 0.2500 | 0.0095 | |
| V1 | 0.37108 (11) | 0.40508 (10) | 0.2500 | 0.0069 | |
| Cl1 | 0.0000 | 0.0000 | 0.0000 | 0.0142 | |
| O1 | 0.4697 (5) | 0.5934 (5) | 0.2500 | 0.0129 | |
| O2 | 0.4809 (5) | 0.3274 (5) | 0.2500 | 0.0164 | |
| O3 | 0.2597 (3) | 0.3541 (4) | 0.0750 (4) | 0.0153 | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Ba1 | 0.01142 (15) | 0.01142 (15) | 0.00687 (19) | 0.00571 (8) | 0.000 | 0.0000 |
| Ba2 | 0.01043 (19) | 0.00921 (18) | 0.00862 (17) | 0.00475 (14) | 0.000 | 0.0000 |
| V1 | 0.0069 (4) | 0.0078 (4) | 0.0065 (4) | 0.0040 (3) | 0.0000 | 0.0000 |
| Cl1 | 0.0128 (6) | 0.0128 (6) | 0.0169 (11) | 0.0064 (3) | 0.0000 | 0.0000 |
| O1 | 0.011 (2) | 0.009 (2) | 0.016 (2) | 0.0026 (16) | 0.0000 | 0.0000 |
| O2 | 0.019 (2) | 0.023 (2) | 0.015 (2) | 0.017 (2) | 0.0000 | 0.0000 |
| O3 | 0.0120 (13) | 0.0238 (16) | 0.0106 (14) | 0.0094 (13) | −0.0030 (12) | −0.0055 (13) |
Geometric parameters (Å, º) top
| Ba1—O1 | 2.751 (3) | Ba2—O2 | 2.974 (5) |
| Ba1—O2i | 2.733 (3) | Ba2—Cl1 | 3.2878 (3) |
| Ba1—O3 | 3.045 (3) | V1—O3 | 1.698 (3) |
| Ba2—O1ii | 2.603 (4) | V1—O2 | 1.722 (4) |
| Ba2—O3iii | 2.699 (3) | V1—O1 | 1.723 (4) |
| Ba2—O3iv | 2.838 (3) | | |
| | | |
| O3—V1—O3v | 106.2 (2) | O3—V1—O1 | 106.78 (14) |
| O3—V1—O2 | 111.95 (14) | O2—V1—O1 | 112.8 (2) |
| Symmetry codes: (i) y, −x+y+1, −z; (ii) −y+1, x−y, z; (iii) y, −x+y, z+1/2; (iv) −x+y, −x, z; (v) x, y, −z+1/2. |


 journal menu
journal menu inorganic compounds
inorganic compounds













 Alert level B
Alert level B
 Alert level C
Alert level C
 Alert level G
Alert level G



