

 journal menu
journal menu





issue contents
November 2013 issue
4th International Symposium on Diffraction Structural Biology (ISDSB2013)
Fukiage Hall, Nagoya, Japan, 26-29 May 2013
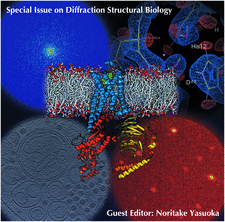
Cover illustration: Images from presentations given at ISDSB2013. Centre: structure of the ![[beta]](/logos/entities/beta_rmgif.gif) -2 adrenergic receptor in complex with its G protein (courtesy of B. Kobilka); ESRF played a major role in the initial GPCR crystal structure determined by the Brian Kobilka Lab, with subsequent structures being obtained with data collected at APS. Top left: an X-ray free-electron laser simulated diffraction pattern with quantum noise for the 70S ribosome (see Tokuhisa et al., pages 899-904). Top right: 2|Fo| - |Fc| neutron-scattering-length map around the active site of RNase A (see Kusaka et al., pages 994-998). Bottom left: slice through the reconstructed tomographic volume of a cryo-EM/ET observation of an RDV-infected NC24 cell (see Miyazaki et al., pages 826-828). Bottom right: time-of-flight neutron diffraction image of transthyretin recorded by iBIX (Yokoyama et al., pages 834-837).
-2 adrenergic receptor in complex with its G protein (courtesy of B. Kobilka); ESRF played a major role in the initial GPCR crystal structure determined by the Brian Kobilka Lab, with subsequent structures being obtained with data collected at APS. Top left: an X-ray free-electron laser simulated diffraction pattern with quantum noise for the 70S ribosome (see Tokuhisa et al., pages 899-904). Top right: 2|Fo| - |Fc| neutron-scattering-length map around the active site of RNase A (see Kusaka et al., pages 994-998). Bottom left: slice through the reconstructed tomographic volume of a cryo-EM/ET observation of an RDV-infected NC24 cell (see Miyazaki et al., pages 826-828). Bottom right: time-of-flight neutron diffraction image of transthyretin recorded by iBIX (Yokoyama et al., pages 834-837).
facility information





diffraction structural biology




 access
access access
access access
access
 access
access access
access access
access
New methodologies at PF AR-NW12A: the implementation of high-pressure macromolecular crystallography
 access
access access
access access
access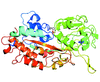 access
access access
access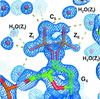 access
access access
access access
access access
access access
access access
access access
access access
access access
access access
access access
access access
access access
access access
access access
access access
access access
access access
access access
access access
access access
access access
access access
access access
access access
access access
access access
access access
access
current events






