

 journal menu
journal menu





issue contents
May 2010 issue

Cover illustration: A fast translation function is used to position a model with underassigned symmetry into the correct unit cell (p. 503). Very near fits to higher symmetry are found for about 2% of PDB entries.
research papers





Open  access
access
access
An X-ray structural model can be reassigned to a higher symmetry space group using the presented framework if its noncrystallographic symmetry operators are close to being exact crystallographic relationships. About 2% of structures in the Protein Data Bank can be reclassified in this way.


The crystal structures of two medium-size proteins (type II dehydroquinases, ∼190 kDa) have been solved using a low-resolution reconstruction obtained by negative-staining EM, a technique that is of wider applicability than cryo-EM.
PDB reference: C. albicans type II dehydroquinase, 3kip
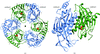
The crystal structure of the RNase PH ring from the exosome of M. thermautotrophicus is described at 2.65 Å resolution.
PDB reference: exosome RNase PH ring, 2wnr
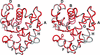
The crystal structure of dehaloperoxidase (DHP) isoenzyme B from the terebellid polychaete A. ornata, which exhibits both hemoglobin and peroxidase functions, has been determined at 1.58 Å resolution.
PDB reference: dehaloperoxidase B, 3ixf

A rich phase diagram of urate oxidase, a protein of high pharmaceutical importance, associated with enhanced polymorphism is revealed.

Structural and inhibition studies of mycobacterial branched-chain aminotransferases were performed with implications for drug design.

Comparative analysis of the prominence of D atoms in neutron and X-ray density maps shows that 1.65 Å resolution neutron diffraction analysis is eightfold more powerful for determining the positions of D atoms than 1.1 Å resolution X-ray diffraction.
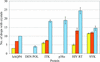
Acoustic liquid handling is employed to deliver crystal seeds in single-digit nanolitre volumes to induce crystallization in sparse-matrix screens while minimizing chemical bias from buffer components in the seed suspension.

Structures of PKC-ι kinase domains have been determined in both apo and ATP-bound forms. The structure of residues 533–551 and the residues around the turn motif in the C-terminal tail are newly defined and help to reveal their roles in ATP binding.

Structure solution using iodide-SAD.

The crystal structures of the apo and holo forms of NADPH-dependent hydroxy(phenyl)pyruvate reductase from C. blumei are reported.

This article presents an overview of protein-engineering methods designed to enhance crystallizability and discusses a number of examples of their successful application.

The structure of human carbonic anhydrase II has been solved with a sulfonamide inhibitor at 0.9 Å resolution. Structural variation and flexibility is seen on the surface of the protein and is consistent with the anisotropic ADPs obtained from refinement. Comparison with 13 other atomic resolution carbonic anhydrase structures shows that surface variation exists even in these highly ordered isomorphous crystals.

The crystal structure of human carbonic anhydrase II with a doubled a axis from that of the usually observed monoclinic cell has been determined and refined to 1.4 Å resolution. The two molecules comprising the asymmetric unit are related by a noncrystallographic translation of ½ along a, but one of the molecules has two alternate orientations related by a rotation of approximately 2°.
PDB reference: human carbonic anhydrase II, 3ks1
short communications

Bacillus subtilis BacB crystallizes in three crystal forms. The differences in the packing between these crystal forms could be rationalized on the basis of metal ions bound on the protein surface that mediate lattice contacts.
PDB reference: BacB, triclinic form, 3h9a
book reviews
Free








