journal menu
journal menu




issue contents
June 2011 issue
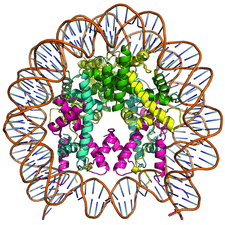
Cover illustration: Human nucleosome containing the histone H3.2 variant (p. 578).
research papers





This article presents the crystal structure of proliferating cellular nuclear antigen from Entamoeba histolytica.
PDB reference: proliferating cellular nuclear antigen, 3p91
The crystal structure of a cellulosomal scaffoldin-borne family 3b carbohydrate-binding module from B. cellulosolvens revealed a Trp-bearing insert that compensates for a Tyr-containing deletion. In contrast to other known structures of family 3 carbohydrate-binding modules, Ca2+ was lacking in the conserved calcium-binding site and a related loop shift placed the Trp in the cellulose-binding planar aromatic strip of the molecule.
PDB reference: carbohydrate-binding module, 2xbt

The spectroscopic and stability properties and X-ray crystal structure of the H87C mutant of the 2Fe–2S ligand mitoNEET are reported. Strikingly, the single point mutation leads to changes in its absorbance and CD spectra and an increase of around sixfold in the stability of the 2Fe–2S clusters over the pH range 5–7. However, the crystal structure of the H87C mutant displays heterogeneity in a few key residues, including the introduced cysteine ligand. Nonetheless, the cluster is highly stabilized from release.
PDB reference: mitoNEET, 3lpq

Structural studies revealed a proline switch as a novel mechanism for the multidrug-resistant nature of multidrug-resistant clinical isolate 769 HIV-1 protease variants.
Open  access
access
access
The X-CHIP (X-ray Crystallography High-throughput Integrated Platform) is a novel microchip that has been developed to combine multiple steps of the crystallographic pipeline from crystallization to diffraction data collection on a single device to streamline the entire process.

Crystal structures of the 6s-98S FIV protease chimera with darunavir and lopinavir bound have been determined at 1.7 and 1.8 Å resolution, respectively.
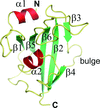
A series of atomic resolution crystal structures of the FK506-binding domain of FKBP51 including a complex with the drug FK506 was solved. The detailed insights into the domain structure and the interaction with FK506 provide an excellent starting point for rational drug design efforts on this molecular chaperone.
Open access
access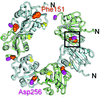
With the aim of forming the `lock-washer' conformation of the origin-binding domain of SV40 large T antigen in solution, using structure-based analysis an intermolecular disulfide bridge was engineered into the origin-binding domain to generate higher order oligomers in solution. The 1.7 Å resolution structure shows that the mutant forms a spiral in the crystal and has the de novo disulfide bond at the protein interface, although structural rearrangements at the interface are observed relative to the wild type.
PDB reference: SV40 large T antigen DNA-binding domain, 3qn2

Analysis of phases derived by phase extension identifies the reciprocal-space volumes that lie close to icosahedral NCS axes as seeding places for incorrect phase solutions.
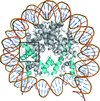
Crystal structures of human nucleosomes containing the histone H3 variants H3.2 or H3.3 have been solved. The structures were essentially the same as that of the H3.1 nucleosome.

Download citation


Download citation
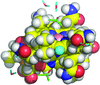
High-resolution crystallographic studies of the hydration of the coenzyme cob(II)alamin have provided hydrogen-bond parameters of unprecedented accuracy for a biomacromolecule.
addenda and errata
Download citation
Download citation
Free
A correction to the article by Nahoum et al. [(2009). Acta Cryst. D65, 832–838].