journal menu
journal menu





issue contents
June 2013 issue
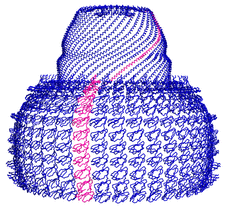
Cover illustration: Overall view of the vault structure refined using DEN protocols (p. 1054). A half-vault structure (in dark blue) with one of the 39 MVP copies forming the particle shown in magenta.
research papers






Open  access
access
access
The crystal structures of the eubacterial translation initiation factor 2 in apo form and with bound GDP and GTP reveal conformational changes upon nucleotide binding and hydrolysis, notably of the catalytically important histidine in the switch II region.

Structural insight into plant programmed cell death mediated by BAG proteins in Arabidopsis thaliana
Crystal structures of the BAG proteins from A. thaliana have been solved at high resolution. Structural and functional data provide evidence of an Hsp70/Hsc70 nucleotide-exchange factor function for these plant BAG proteins.
PDB references: AtBAG1 BD, 4hwc; AtBAG2 BD, 4hwd; AtBAG3 BD, 4hwf; AtBAG4 BD, 4hwh; AtBAG1 UBD–NBD complex, 4hwi
An analytical approach is presented to identify and resolve several distinct structural species dynamically mixed in multiple crystallographic data sets.
The X-ray crystal structure of geodin is reported.
PDB reference: geodin, 4iau

The crystal structures of two circularly permuted streptavidins probe the role of a flexible loop in the tight binding of biotin. Molecular-dynamics calculations for one of the mutants suggests that increased fluctuations in a hydrogen bond between the protein and biotin are associated with cleavage of the binding loop.
High-resolution powder X-ray diffraction data have been collected to reveal the T6 hexameric insulin form and a novel approach for refining protein structures using powder diffraction data is presented.
PDB reference: T6 hexameric insulin, 4idw

The Glu43Ala/Phe81Ala mutation in binase prevents dimerization of the enzyme and improves its catalytic activity and cytotoxicity.
PDB reference: binase, Glu43Ala/Phe81Ala mutant, 4haa
The 2.4 Å resolution crystal structure of the H. marismortui large ribosomal subunit has been re-refined; the structures of r-proteins P0 and P1 were visualized in part and r-protein LX and helix 76 of the 23S rRNA were localized.
The yellow fluorescent protein phiYFPv with improved folding has been developed from the spectrally identical wild-type phiYFP found in the marine jellyfish Phialidium.
PDB reference: phiYFPv, 4he4
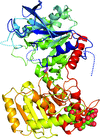
The crystal structure of starch synthase I from barley was refined to 2.7 Å resolution. It includes a regulatory disulfide and a bound oligosaccharide. Activity assays were performed on several mutants.
PDB reference: starch synthase I, 4hln
Open access
access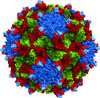
The crystallographic structure of TrV shows specific morphological and functional features that clearly distinguish it from the type species of the Cripavirus genus, CrPV.
PDB reference: TrV, 3nap

The VLD approach has been integrated into a molecular-replacement pipeline for automatic protein crystal structure solution.
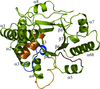
Structures of the EAL domain of the E. coli direct oxygen sensor show the active site in a nonproductive conformation that has implications for the regulatory mechanism.

Here, the re-refinement of the structure of the vault particle by incorporating the high-resolution information available for the R1–7 domains, using the deformable elastic network (DEN) approach and maintaining strict 39-fold noncrystallographic symmetry is reported.
PDB reference: Re-refinement of the vault ribonucleoprotein particle, 4hl8


A computer simulation was created for a modulated protein structure along with structure factors in a periodic supercell and in superspace for the purpose of developing and validating software modifications that will be used to solve and refine modulated protein crystals.
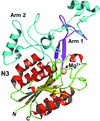
The crystal structure of a 75 kDa central fragment of GBS104, a tip pilin from the 2063V/R strain of Streptococcus agalactiae (group B streptococcus; GBS), is reported.
Open access
access
The putative methyltransferase CmoA is involved in the nucleoside modification of transfer RNA. X-ray crystallography and mass spectrometry are used to show that it contains a novel SAM derivative, S-adenosyl-S-carboxymethyl-L-homocysteine, in which the donor methyl group is replaced by a carboxymethyl group.
PDB reference: CmoA, 4iwn

The X-ray crystal structure of an outer surface protein BBA64 from the Lyme disease agent B. burgdorferi which is necessary to cause the disease is reported and compared with that of the homologous protein CspA from B. burgdorferi.
PDB reference: BBA64, 4aly

ps-2-HAD catalyses both L- and D- substrates, showing similar overall folding to L-HADs but completely different to the only structurally characterised DL-2-HAD enzyme.
PDB reference: ps-2-HAD, 3vay
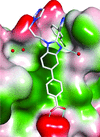
The bound structure of a novel HIV-1 capsid-assembly inhibitor describes a unique inhibitor-binding site on the capsid N-terminal domain. Moreover, use of this inhibitor as a crystallization tool greatly improved the success rate of capsid N-terminal domain ternary cocrystallizations via compound-mediated synthetic dimerization.
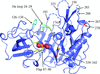
Surface-residue mutagenesis and cocrystallization with Fab fragments, Fynomers or Xaperones were used to obtain the first high-resolution crystal structures of BACE2. These map the conformational space accessible to this potential drug target.
This study underscores the plasticity of the multidrug-binding pocket in MarR proteins and may help for the understanding of the antimicrobial resistance mechanism in pathogens.
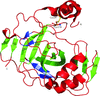
The crystal structure of SspCA, a novel `extremo-α-carbonic anhydrase', is described, providing an elucidation of the factors responsible for its function at high temperature.
PDB reference: SspCA, 4g7a
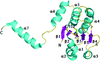
This study presents crystal structures of the catalytic domain of human DUSP26 and a catalytically inactive mutant at 1.67 and 2.20 Å resolution, respectively.
New programs for automated nucleic acid structure refinement, NAFIT and NABUILD, are described. They fit and extend the nucleic acid model and work with the automated iterative refinement system LAFIRE.
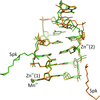
Ultrahigh-resolution crystal structures of metal complexes of self-complementary Z-DNA with the sequence d(CG)3 revealed different coordination patterns for Mn2+ (octahedral) and Zn2+ (octahedral and tetrahedral) cations. The complete sperminium cation is visible in electron density in the Mn2+ structure, while in the Zn2+ structure it is partly disordered in unison with fragments of the DNA molecule.
addenda and errata
Free

Free