journal menu
journal menu





issue contents
September 2013 issue
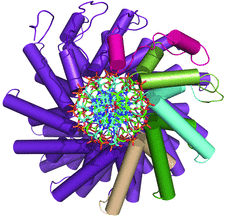
Cover illustration: Cartoon representation of the TALE AvrBs3-DNA complex structure. The figure shows the protein helical repeats wrapping around the double strand DNA. Each repeat contains the dipeptides responsible of specific DNA recognition (p. 1707). The initial repeats are colored in red, lime, cyan, green and beige to indicate the initial DNA domain building blocks. The specific protein-DNA interactions involve only the coding DNA strand colored in green.
research papers






This study represents the first crystal structure of bi-functional ribB/ribA enzyme from Mycobacterium tuberculosis.
PDB reference: DHBPS/GCHII, 4i14
The crystal structures of the GluA2 ligand-binding domain in complex with the positive allosteric modulator CX516 and its methyl-substituted analogue Me-CX516 show that their binding modes are similar to those of aniracetam and CX614. This supports that CX516 affects receptor deactivation. The structures also show that there is limited space for substitutions at the piperidine ring of CX516.

The first structure of the N-terminal domain of the Mg2+ channel Mrs2 in a monomeric form and a pentameric model of Mrs2 generated using the prokaryotic homologue CorA are presented. Gating and metal-sensing residues were proposed based on the model and were validated using mutational and functional studies in vivo.
PDB reference: Mrs248–308, 3rkg

Expression profiling of the three homodimeric (prototype) chicken galectins (CG-1A, CG-1B and CG-2) has raised evidence of distinct functionalities, explaining the interest in a detailed crystallographic analysis of CG-2. Marked differences are found in the ligand-binding site and in the contact pattern within the homodimer interface, underlying a characteristic orientation of the two subunits. Notably, a distinctive trimer of dimers that is unique in all galectin crystal structures reported to date forms the core unit of the crystallographic assembly.
PDB reference: chicken galectin 2, 2ymz

The crystal structure of a novel C-terminal dimerization domain of the cullin-3 adaptor SPOP is reported. The dimerization interfaces of the SPOP BTB and C-terminal domains act in tandem to generate high-order oligomers that enhance the ubiquitination of substrates.
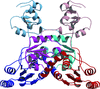
Structural studies of quinolinate synthase suggest a model for the enzyme–substrate complex and an enzyme–intermediate complex with a [4Fe–4S] cluster.
PDB reference: quinolinate synthase, 4hhe
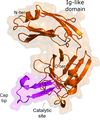
This work reports the crystal structures of the L,D-transpeptidase LdtMt1 from M. tuberculosis in a ligand-free form and in complex with the carbapenem imipenem.
Open  access
access
access
The crystal structure of the AvrBs3–DNA complex is reported.
PDB reference: AvrBs3–DNA complex, 2ypf
Open access
access
The high-resolution crystal structures of apo and peptide-bound XIAP BIR2 are presented and compared with BIR3 structures to understand their selectivity. This crystal system can be used to determine the structures of BIR2–inhibitor complexes.

The crystal structure of Est25, a bacterial homologue of hormone-sensitive lipase (HSL), was determined at 1.49 Å resolution. The biochemical properties of Est25 were investigated to clarify the functional capacities of HSL and the potential applications of Est25 in biotechnology.
PDB reference: Est25, 4j7a
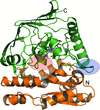
Crystallographic, functional and single-molecule studies of aminopeptidase PepS have been presented. Substrate length selectivity imposed by the substrate binding hole is a key in the regulation of the catalytic activity along with residue specificity and active-site repositioning.
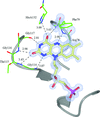
Diffraction analysis of E. coli WrbA crystallized in the presence of FAD yields the highest resolution structure yet reported, but with FMN bound, Met10 oxidized, and a propeller-twisted isoalloxazine ring.
PDB reference: WrbA, 3zho

Crystal structure analyses of the monooxygenase TetX in complex with tigecycline and minocycline support the observation that all clinically available tetracycline antibiotics can be inactivated by enzymatic hydroxylation. Molecular-dynamics simulations of dioxygen diffusion and experimental xenon-binding sites in TetX are compared with respect to putative dioxygen-binding cavities and dioxygen-diffusion pathways.

Magnesium bound to T. brucei pyruvate kinase in the presence of the allosteric activator fructose bisphosphate, but in the absence of the substrate phosphoenolpyruvate, is sequestered in a previously undescribed `priming' position adjacent to the active site. In this way, the enzyme is kept in a conformation with a high affinity for the substrate.
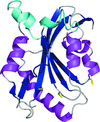
The crystal structure of the human actin-regulating protein cofilin 1, and comparisons with yeast, Chromalveolata and plant homologues that reveal conformational changes required for actin binding, are reported.
PDB reference: human cofilin 1, 4bex
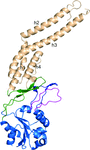
The crystal structure of the intact substrate-recognition domain of a LonC protease shows a large helical hairpin extension (HHE). The HHEs form a basket-like structure on top of the degradation chamber of the LonC protease. The HHE of LonC shares structural and functional similarity to the α-helical tentacle of the periplasmic chaperone Skp.
PDB reference: N-terminal domain of MtaLonC, 4fwv
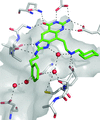
The simultaneous filling of the ribose-33 and ribose-34 pockets at the active site of Z. mobilis tRNA–guanine transglycosylase was investigated using a series of bifunctionalized lin-benzopurine inhibitors. A strong influence of the crystallization protocol on the solvation pattern and the residual mobility of the bound ligands was observed in the cocrystal structures.
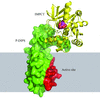
The T. maritima inositol-1-phosphate cytidylyltransferase structure provides insights into its mechanism of forming CDP-inositol and its interaction with the membrane-bound enzyme in the next step of the biosynthesis of the unusual osmolyte di-myo-inositol-1,1′-phosphate in hyperthermophiles.
PDB reference: inositol-1-phosphate cytidylyltransferase, 4jd0
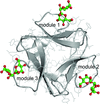
The fully mature anti-HIV lectin actinohivin in complex with the target α(1–2)mannobiose moiety of the high-mannose-type glycans attached to HIV-1 gp120 has been crystallized into two new forms without packing disorder and their X-ray structures reveal the detailed interaction geometry for specific binding to the target HIV-1.
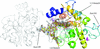
The first structure of a human β-galactoside α-2,6-sialyltransferase I was determined by SIRAS phasing using an iodide soak for derivatization. An elongated glycan from a crystallographic neighbour binds to the active site, mimicking a substrate complex. An analysis of the substrate interactions and a comparison with other sialyltransferases allowed the modelling of a Michaelis complex and conclusions on the catalytic mechanism.
Open access
access
A new crystal-mounting method has been developed that involves a combination of controlled humid air and polymer glue for crystal coating. This method is particularly useful when applied to fragile protein crystals that are known to be sensitive to subtle changes in their physicochemical environment.
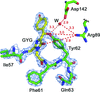
The crystal structure of the novel red emitting fluorescent protein from lancelet Branchiostoma lanceolatum (Chordata) revealed an unusual five residues cyclic unit comprising Gly58-Tyr59-Gly60 chromophore, the following Phe61 and Tyr62 covalently bound to chromophore Tyr59.
short communications

mrtailor is a tool to improve PDB files for the generation of external restraints for refinement.
addenda and errata
Free

A correction is made to the article by Harding [(2002) Acta Cryst. D58, 872–874].