

 journal menu
journal menu




issue contents
February 2014 issue
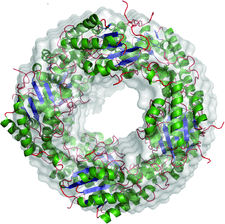
Cover illustration: When crystallography meets SAXS. The structure of the Axe2 octamer, as has been determined by X-ray crystallography, and the molecular envelope of the same octamer, as obtained from independent SAXS experiments in solution (p. 261). The excellent fit of the two images demonstrates that the crystallographic octamer is the relevant biological form of the protein.
research papers





Vapour soaking of crystals of the R. rhodochrous haloalkane dehalogenase variant DhaA31 with 1,2,3-trichloropropane at room temperature and pH 6.5 facilitated the production of a protein–substrate complex. Comparison of the substrate-free DhaA31 structure with that of the DhaA31–substrate complex revealed two alternative conformations of the nucleophilic residue Asp106 and a possible pathway for halide release, while comparison of DhaA31 with wild-type DhaA revealed reduced size and solvent-accessibility of the active site.
The crystal structure of the α subdomain of the Lon protease from Brevibacillus thermoruber is presented, together with biochemical data, and the DNA-binding mode is delineated, showing that Arg518, Arg557 and Arg566 play a crucial role in DNA binding.
PDB reference: α subdomain of Bt-Lon, 4git

The structures of human squalene synthase and its mutants in complex with several substrate analogues and intermediates coordinated with Mg2+ or Mn2+ stepwise delineate the biosynthetic pathway.
This work reports the structural basis of the highly efficient catalysis of most PDF enzymes by analyzing the impact of the two main substitutions of animal mitochondrial PDFs which cause unusually weak rate.
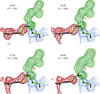
Refinement and analysis of four structures with various data resolution cutoffs suggests that at present there are no reliable criteria for judging the diffraction data resolution limit and the condition I/σ(I) = 2.0 is reasonable. However, extending the limit by about 0.2 Å beyond the resolution defined by this threshold does not deteriorate the quality of refined structures and in some cases may be beneficial.

The crystal structure of the serine acetyl-xylooligosaccharide esterase from G. stearothermophilus (Axe2) has been determined by SAD techniques, allowing full structural analysis of the selenomethionine derivative Axe2-Se (at 1.70 Å resolution), the wild type Axe2-WT (1.85 Å) and the catalytic mutant Axe2-S15A (1.90 Å). The structures show that the enzyme forms a unique octameric torus built of two staggered cyclic tetramers, with four pairs of catalytic triads facing the central cavity. This octameric torus is further confirmed by gel-filtration, TEM and SAXS results.
Open  access
access
access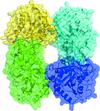
The structure of a bacterial M14-family carboxypeptidase determined exploiting microfocus synchrotron radiation and highly automated refinement protocols reveals its potential to act as a polyglutamylase.
PDB reference: carboxypeptidase, 4b6z
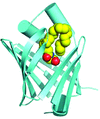
FABP5 was recently found to intracellularly transport endocannabinoid signaling lipids. The structures of FABP5 complexed with two endocannabinoids and an inhibitor were solved. Human FABP5 was found to dimerize via a domain-swapping mechanism. This work will help in the development of inhibitors to raise endocannabinoid levels.
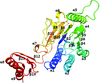
In this study, crystal structures of the E. coli Mre11 homologue SbcD and its Mn2+ complex are reported and an ssDNA-binding model is proposed for SbcD that differs from those of other Mre11 proteins, providing insight into the catalytic mechanism of the repair of DNA double-strand breaks by the Mre11 complex.
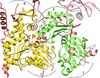
The crystal structure of a complex of full-length Arp7 and Arp9 was determined that shows that they form a heterodimer, in contrast to the monomeric units observed for other actin-related proteins. Therefore, the dimeric Arp7–Arp9 form that purportedly exists in the SWI/SNF and RSC chromatin-remodelling factors exists independently of the binding of other proteins in these megadalton complexes.
PDB reference: Arp7–Arp9 complex, 3wee

Specifically deuterated phospholipid bilayer nanodiscs have been developed which give a minimal contribution to neutron scattering data when used in 100% D2O. These provide an optimal platform for low-resolution structural determination of membrane proteins and their complexes in solution.
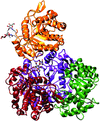
This study describes the three-dimensional structure of the endogenous glycosylated allergen Hev b 2 (endo-β-1,3-glucanase), which exhibits three post-translational modifications that form a patch on the surface of the molecule that is proposed to be an allergenic IgE epitope.

This study presents the crystal structure of a ∼320 Å long protein fiber generated by in-frame extension of its repeated helical coiled-coil core.
PDB reference: gp26-2M, 4lin
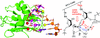
Selenium-derivatized oligonucleotides may facilitate phase determination and high-resolution structure determination for protein–nucleic acid crystallography. The Se atom-specific mutagenesis (SAM) strategy may also enhance the study of nuclease catalysis.
PDB reference: RNase H–RNA/DNA complex, 3twh
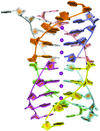
The high-resolution crystal structure of an all-LNA G-quadruplex provides important information on the role of this modified residue on structural and physical properties of quadruplexes. The highly packed assembly observed in the crystal suggests a way by which quadruplexes can be arranged in crowded environments and can be used in nanotechnological applications.
PDB reference: all-LNA quadruplex, 4l0a
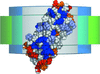
A hybrid approach to the analysis of small-angle scattering data is presented, allowing determination of the orientation of bacteriorhodopsin reconstituted into a phopholipid nanodisc. The method is applicable to similar systems.
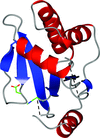
The Salmonella effector protein GtgE functions as a cysteine protease to cleave a subset of the Rab-family GTPases and to prevent delivery of antimicrobial agents to the Salmonella-containing vacuole.
PDB reference: GtgE, 4mi7
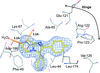
Co-crystals of SKF86002, a small kinase inhibitor, and various kinases were distinguishable by their strong fluorescence, and the crystals lost their fluorescence when a compound bound competitively to the kinase ATP-binding site and displaced the fluorescent SKF86002. SKF86002 binds a wide variety of kinases and thus would be a useful crystal marker, crystal stabilizer and marker for screening new kinase inhibitors and identifying ligand co-crystals for structural analysis.
PDB references: Pim1–SKF86002, 4ll5; Pim1–quercetin, 4lmu; Pim1–compound 1, 4lm5; HCK–SKF86002, 4lud; HCK–A-419259, 4lue
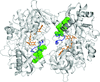
The crystal structure of choline oxidase in complex with the physiological product glycine betaine allows comparison with previous mechanistic studies of choline oxidase and highlights a distinct conformation of a loop at the dimer interface which is suggested to regulate substrate access to the active site.
PDB reference: choline oxidase, complex with glycine betaine, 4mjw

The 2.2 Å resolution neutron structure of the xylose isomerase E186 mutant with cyclic glucose bound at the active site reveals an extended hydrogen-bond network that connects conserved residues and suggests a catalytic role for Lys289.
PDB reference: cyclic glucose-bound xylose isomerase, 4lnc
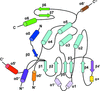
A family-wide structural library of human DUSPs was constructed, which provides a basis for the understanding of phosphorylation-mediated signal transduction and the development of therapeutics.
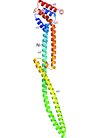
LegC3 is an effector secreted by Legionella pneumophila, and is believed to act by inhibiting vacuolar fusion. The function of LegC3 has been shown to be associated primarily with the N-terminal domain, the structure of which has been determined here.
PDB reference: N-terminal domain of LegC3, 4mu6

S-Adenosylmethionine synthetase catalyzes the production of the major biological methyl donor, which is also the source for moieties that participate in a range of different group transfer reactions. The structure of the enzyme form obtained from Campylobacter jejuni provides explanations for its higher catalytic activity, altered substrate specificity and unusual dimeric quaternary assembly.
PDB reference: S-adenosylmethionine synthetase, 4le5
Open access
access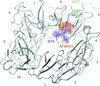
The identification of the first small-molecule ligand of the neuronal receptor sortilin and structure determination of the receptor–ligand complex are reported.
PDB reference: sortilin–AF40431 complex, 4msl
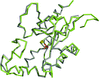
High-resolution crystal structures of mitochondrial and cytosolic 5′-deoxyribonucleotidases with active-site phosphate ions were used to estimate phosphate protonation and to investigate differences in their active sites. These findings were applied to the design of a putative specific inhibitor.
Open access
access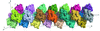
With the implementation of a molecular-replacement likelihood target that accounts for translational noncrystallographic symmetry, it became possible to solve the crystal structure of a protein with seven tetrameric assemblies arrayed translationally along the c axis. The new algorithm found 56 protein molecules in reduced symmetry (P1), which was used to resolve space-group ambiguity caused by severe twinning.

Conformational differences between myoglobin structures are studied. Most structural differences in whale myoglobin beyond the uncertainty threshold can be correlated with a few specific structural factors. There are always exceptions and a search for additional factors is needed. The results might have serious implications for biological insights from conformational differences.
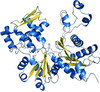
The crystal structure of the heat-stable archaeal actin, crenactin, is revealed and atomic-level details are presented and compared with those of eukaryotic actin and bacterial MreB.
PDB reference: crenactin, 4bql
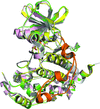
Four crystal structures of S. cerevisiae CK2α (scCK2α) with different nucleotide co-substrates and coordinated divalent cations indicated that scCK2α possesses dual co-substrate specificity, possible multiple nucleotide–divalent cation binding modes and a possible ADP/GDP-release pathway by means of conformational change of β1/G-loop/β2. The presence of a unique insertion loop in scCK2α enhanced the catalytic efficiency of the enzyme by stabilizing the open conformation of the catalytic site.
PDB references: scCK2α–ATP–Mg2+, 4mwh; scCK2α–AMPPN, 4jqe; scCK2α–GMPPNP–Mg2+, 4jr7; scK2α–GMPPNP–Mn2+, 4lfi
Open access
access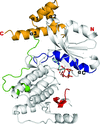
The structure of a crystal of MPK38 (T167E), which consists of a kinase domain and a UBA domain, in complex with AMP-PNP is reported at 2.4 Å resolution. The structure indicates that the activation of MPK38 is induced by the UBA linker restraining the motion of the αC helix and by phosphorylation of Thr167 stabilizing the activation loop.
PDB reference: MPK38 (T167E), complex with AMP-PNP, 4bfm

The expression of carbohydrate-active proteins from C. thermocellum is proposed to be regulated by a σI/anti-σI (RsgI) mechanism that is sensitive to extracellular carbohydrates. Here, the structures of three putative RsgI carbohydrate-sensing modules belonging to the family 3 carbohydrate-binding modules are described.

The crystal structure of the Cmr1 subunit of the Cmr interference complex reveals a single-stranded RNA-binding site and an associated ribonuclease activity.
PDB reference: Af Cmr1, 4l6u
Open access
access
The principles of the Bürgi–Dunitz nucleophilic approach necessitate structural rearrangements in Schiff-base-forming enzymes. Presented as a case study, substrate- and reaction intermediate-bound crystal complexes reveal that substrate conformational changes achieve the required rearrangements in transaldolase Schiff-base formation.

The reduction reactions of different substrates by azoreductases have been investigated based on the crystal structures of AzrA and of AzrC–inhibitor/substrate complexes. The structures of the AzrC–substrate complexes present two different manners of binding, suggesting two types of reduction reaction depending on the type of substrate.

A nitrate molecule was found in both the open and closed form of the catalytic site of PTP1B. This complex was modelled using quantum mechanics and compared to the crystal structure.
PDB reference: protein tyrosine phosphatase 1B, 4bjo
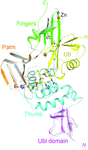
The crystal structure of the papain-like protease (PLpro) C112S mutant in complex with ubiquitin offers insight into the molecular basis of the catalytic mechanism of PLpro and its substrate binding. An N-cyclohexyl-2-aminethanesulfonic acid molecule in the active site suggests a possible approach to inhibition.
PDB reference: PLpro C112S mutant, complex with ubiquitin, 4m0w
Open access
access
High-resolution crystal structures together with mutational analysis and transient kinetics experiments were utilized to understand nucleotide sensing and the regulation of the ATPase cycle in an AAA+ molecular motor.

Download citation


Download citation

Structural snapshots of the L-serine dehydratase catalytic reactions of a PLP-dependent enzyme were determined by X-ray crystallography. PLP cofactor catalyzed a series of reactions with active conformational changes and its catalytic role was confirmed by high-level quantum-mechanical calculations.
Open access
access
The binding modes of acivicin, a classical and an electrophilic active-site-directed glutamate analogue, to bacterial γ-glutamyltranspeptidases were found to be diverse.





