

 journal menu
journal menu




issue contents
June 2014 issue
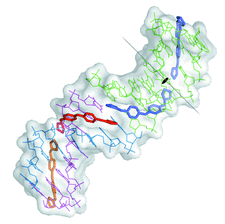
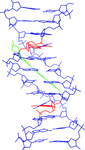
Cover illustration: The crystal structure of the complex of the DNA duplex d(AAAATTTT)2 with the dicationic drug 4,4'-bis(imidazolinylamino)diphenylamine (CD27) (p. 1614).
research papers






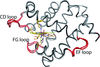
A high resolution crystal structure of the nisin leader peptidase NisP is reported, which revealing a C-terminal autocleavage activity.
PDB reference: NisP, 4mzd
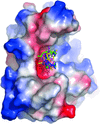
A set of seven caged gadolinium complexes were shown to be excellent compounds for introducing anomalous scatterers into protein crystals for de novo anomalous phasing. Their high phasing power and versatile binding modes make them highly suitable as a heavy-atom screen.
Open  access
access
access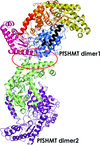
The crystal structure of P. falciparum SHMT revealed snapshots of an intriguing disulfide/sulfhydryl switch controlling the functional activity.
PDB reference: serine hydroxymethyltransferase, 4o6z

The crystal structure of the bacterial diterpence cyclase CotB2 is presented. Mutation of residues with plasticity in the active site leads to altered products.
Open access
access
The idea of attacking the phase problem by crowdsourcing is introduced. Using an interactive, multi-player, web-based system, participants work simultaneously to select phase sets that correspond to better electron-density maps in order to solve low-resolution phasing problems.
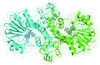
The first structural complexes of the ferric ion-SAM-dependent C-methyltransferase MppJ are reported, whereon MppJ was engineered performing unforeseen Lewis acid-assisted hydration and/or O-methyltransfer reactions to form novel stereospecific compounds.

The crystal structure of the C-terminal phosphotransferase domain of the bifunctional aminoglycoside acetyltransferase–phosphotransferase AAC(6′)-Ie-APH(2′′)-Ia has been determined to 2.3 Å resolution. The enzyme was crystallized as the Mg2GDP binary complex.
PDB reference: APH(2′′)-Ia, 4ork
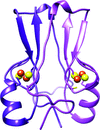
NAF-1 has been shown to be related with human health and disease, is upregulated in epithelial breast cancer and suppression of its expression significantly suppresses tumor growth. It is shown that replacement of the single His ligand with Cys resulted in dramatic changes to the properties of its 2Fe-2S clusters without any global crystal structural changes.

The widely used 2003 Matthews probability calculator predicting the most probable number of molecules in the asymmetric unit as a function of resolution has been updated with 2013 data, and a new parameter-free kernel density estimator has been implemented. Analysis of multiple parameters confirms that solvent content remains the primary and dominating parameter determining the diffraction limit (resolution) of protein crystals.

Download citation


Download citation

Structural comparison among three crystal forms of the measles virus phosphoprotein multimerization domain unveiled significant discrepancies in the quaternary structure. Results indicate that coiled coils are less rigid than previously thought and challenge to some extent the assumption according to which coiled-coil structures can be reliably predicted from the amino-acid sequence.

Two crystal structures of calmodulin in complexes with the corresponding binding regions of the death receptors p75NTR and Fas are reported.
Open access
access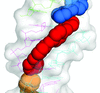
New features of an antiprotozoal DNA minor-groove binding drug, which acts as a cross-linking agent, are presented. It also fills the minor groove of DNA completely and prevents the access of proteins. These features are also expected for other minor-groove binding drugs when associated with suitable DNA targets.
PDB reference: d(AAAATTTT)2–CD27, 4ocd
Open access
access
The crystal structure of the helicase domain of the human spliceosomal DEAD-box protein Prp28 was solved by SAD. The binding of ADP and ATP by Prp28 was studied biochemically and analysed with regard to the crystal structure.
PDB reference: Prp28, 4nho
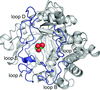
Crystal structures of the β-glucosidase from the fungus H. insolens provided new insights into the structural determinants for glucose tolerance in β-glucosidases.
Mutants have been produced that reduce the volume of the large internal cavity of murine neuroglobin. Their structural and functional characterization sheds light on the role of the cavity itself and of the structural transitions of the haem upon binding.

The crystal structure of the wound-induced leucine aminopeptidase from tomato was determined to 2.20 Å resolution by the molecular-replacement method. Structural analysis and modeling reveal the structural basis of substrate binding.
PDB reference: wound-induced leucine aminopeptidase, 4ksi
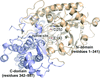
The 1.8 Å resolution crystal structure of a novel maltose-forming α-amylase (PSMA) recently found in the hyperthermophilic archaeon Pyrococcus sp. ST04 is reported.
PDB reference: maltose-forming α-amylase, 4cmr
The O6-carboxymethylguanine residue identified in DNA from human colon tissue, which has been linked to diets high in red and processed meats, is revealed to form a base pair with a thymine residue of a new high-wobble type in addition to the Watson–Crick type.
Open access
access
Crystals of human CRMP-4 showed severe lattice-translocation disorder. Intensities were demodulated using the so-called lattice-alignment method and a new more general method with simplified parameterization, and the structure is presented.
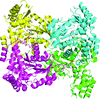
The crystal structures of A. jandaei L-allo-threonine aldolase and a mutant were refined to 2.59 and 2.50 Å resolution, respectively. The structural basis of the substrate specificity and stereoselectivity has been elucidated.
Open access
access
The structure of the human C5aR antagonist, C5a-A8, reveals a three-helix bundle conformation similar to that observed for human C5a-desArg, whereas murine C5a and C5a-desArg both form the canonical four-helix bundle. These conformational differences are discussed in light of the differential C5aR activation properties observed for the human and murine complement anaphylatoxins across species.
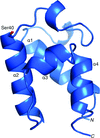
The high-resolution crystal structure of a free-standing carrier protein from Acinetobacter baumannii that belongs to a larger NRPS-containing operon, encoded by the ABBFA_003406–ABBFA_003399 genes of A. baumannii strain AB307-0294, that has been implicated in A. baumannii motility, quorum sensing and biofilm formation, is presented.
PDB reference: NRPS PCP domain, 4hkg

Binding of cAMP and cGMP to the global transcription factor CRP results in different allosteric transitions of the protein. cGMP binding induces an inhibitory conformational change of CRP.
Open access
access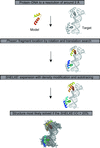
The structure solution of DNA-binding protein structures and complexes based on the combination of location of DNA-binding protein motif fragments with density modification in a multi-solution frame is described.
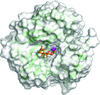
The structure of human carbonic anhydrase II in complex with cholate has been determined to 1.54 Å resolution. Elucidation of the novel inhibition mechanism of cholate will aid in the development of a nonsulfur-containing, isoform-specific therapeutic agent.
PDB reference: human carbonic anhydrase II–cholate complex, 4n16
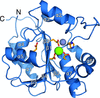
The crystal structure of the Lymphocytic choriomeningitis virus (LCMV) nucleoprotein C-terminal domain adopts the fold of an exonuclease and provides an important structural template for the study of this prototypic arenavirus.
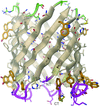
The 1.9 Å resolution structure of a 12-stranded specific porin is presented. Two tracks of basic residues at the pore lining probably facilitate translocation of acidic oligosaccharides.
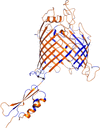
The cloning, purification and crystallization of the E. coli BamA β-barrel domain including one POTRA domain is reported. The crystal structure allows detailed insights into the mechanism of outer membrane protein biogenesis in Gram-negative bacteria.
PDB reference: BamA, 4c4v





