journal menu
journal menu





issue contents
February 2015 issue
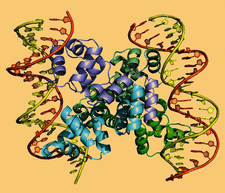
Cover illustration: The structure of an example of a new class of controller proteins (C.Csp231I) in complex with its 21 bp DNA-recognition sequence (Shevtsov et al., p. 398). The two dimers present in the asymmetric unit of the C2 crystal are shown as ribbon diagrams with their respective DNA operators.
research papers







The storage polysaccharide of brown algae is laminarin, a β-1,3-glucan with occasional β-1,6 branches. The catalytic module of the β-glucanase ZgLamC from the marine bacterium Z. galactanivorans has been characterized. Notably, the structure of an inactive mutant of ZgLamC in complex with a thio-β-glucan analogue suggests specific recognition of the branched domains of laminarin.
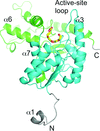
X-ray crystal structures of LDHA in complex with two quinoline-based inhibitors show the inhibitors bound to a site overlapping the NADH-binding site. Apo, oxalate/NADH-bound and oxalate/kanamycin-bound LDHA structures are also reported.

The crystal structure of a synthetic consensus pentatricopeptide repeat protein displays helical disorder. The structure provides support for the utility of synthPPR as a backbone for designing bespoke RNA-binding proteins.
PDB reference: synthPPR3.5, 4ozs
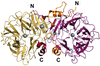
Structural, functional and phylogenetic analyses reveal enzyme diversity within family 117 glycoside hydrolases, which remove terminal α-1,3-linked 3,6-anhydro-L-galactose from neoagaro-oligosaccharides produced by β-agarases. A product complex with the unique β-3,6-anhydro-L-galactose provides strong crystallographic evidence for an inverting mechanism in this family of enzymes.
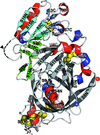
The crystal structure of the 45 kDa γ-conglutin from lupin seeds was determined at 2 Å resolution, showing that in contrast to the tetrameric structure of another 7S basic globulin from a legume plant (soybean), it forms a ring-like hexamer.
PDB reference: γ-conglutin, 4pph
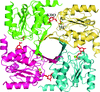
The crystal structure of KdsC from M. catarrhalis reveals an intersubunit water tunnel proposed to provide a water molecule at the active site for catalysis.
Open  access
access
access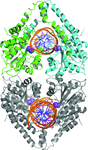
A time-resolved study using the freeze-trap method elucidates the mechanism of the DNA-cleaving reaction of HindIII.
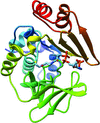
The structure of Mesorhizobium loti arylamine N-acetyltransferase 1 (NAT1) in complex with coenzyme A provides further understanding of the mode of binding of the cofactor in this family of enzymes.
Open access
access
Determination of the nitrogenase MoFe protein from C. pasteurianum at 1.08 Å resolution and comparison to its distinct ortholog from A. vinelandii at atomic resolution reveals conserved structural arrangements that are significant to the function of nitrogenase.
PDB reference: MoFe protein, 4wes
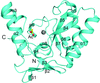
The structure of the L,D-carboxypeptidase DacB from S. pneumoniae provides novel insights into substrate recognition and catalysis in the metallopeptidase M15B subfamily.
PDB reference: DacB, 4nt9

Algorithms are proposed which enable the calculation of the time-accumulated diffraction intensities for a biological macromolecule with time-dependent atomic scattering factors at a computational cost comparable with that for the conventional time-independent case.

Low-accuracy de novo models can be used successfully for ab initio phasing by the reassembly of molecular-replacement solutions of individual fragments derived from these de novo models.

Structural and functional studies of an NAD-dependent formate dehydrogenase from Thiobacillus sp. KNK65MA (TsFDH) revealed that it is an efficient CO2-reducing biocatalyst.
PDB reference: NAD-dependent formate dehydrogenase, 3wr5

Using the crystal structures of complexes of jacalin with α- and β-galactosides, supplemented by thermodynamic data, it has been demonstrated that the low affinity of the lectin for β-galactosides is caused by distortion of the ligand and not by unfavourable protein–ligand interactions.

The crystal structure and sequence analysis of the protein SAV1646 from S. aureus identify it as a member of a new subfamily of `ribosome-associated' proteins. Their hypothetical function is discussed.
PDB reference: SAV1646, 2p92
Open access
access
Two ab initio modelling programs solve complementary sets of targets, enhancing the success of AMPLE with small proteins.
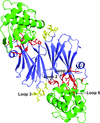
The 1.24 Å resolution crystal structure of the periplasmic i-type lysozyme inhibitor from Aeromonas hydrophila (PliI-Ah) in complex with the i-type lysozyme from Meretrix lusoria is reported.
PDB reference: PliI–lysozyme complex, 4pj2
Open access
access
This paper describes a set of tools allowing experimentalists insight into the variation present within large serial data sets.
Open access
access
Special methods are required to interpret sparse diffraction patterns collected from peptide crystals at X-ray free-electron lasers. Bragg spots can be indexed from composite-image powder rings, with crystal orientations then deduced from a very limited number of spot positions.
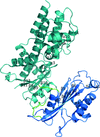
A biochemical and structural study of Arabidopsis hexokinase 1 has shown that a glucose-induced conformational change serves as a signalling event.
Comparison of the structures of RNA complexes of wild-type and mutant variants of TthL1 and analysis of the kinetics of RNA binding to the protein show that complex formation restores the distorted RNA-binding surface of the mutated protein and that the affinity of the protein for RNA cannot be correlated with the number of intermolecular contacts and hydrogen bonds.
High-throughput, room-temperature serial crystallography using a slow stream is demonstrated at a synchrotron source. The data quality is sufficiently good to allow de novo phasing.
Open access
access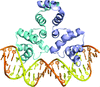
The structure of the new class of controller proteins (exemplified by C.Csp231I) in complex with its 21 bp DNA-recognition sequence is presented, and the molecular basis of sequence recognition in this class of proteins is discussed. An unusual extended spacer between the dimer binding sites suggests a novel interaction between the two C-protein dimers.
Open access
access
A high-resolution structure of a noncanonical α-mannanase relevant to human health and nutrition has been solved via heavy-atom phasing of a selenomethionine derivative.
addenda and errata
Open access
access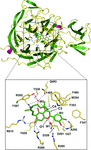
The article by Lee et al. [(2014) Acta Cryst. D70, 1357–1365] is corrected.