

 journal menu
journal menu





issue contents
April 2018 issue
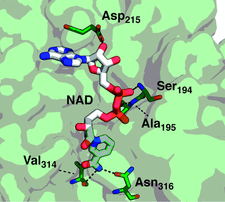
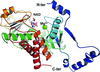
Cover illustration: Phytoplasmas are phytopathogenic bacteria that produce devastating effects in a wide variety of plants. The crystal structure of the malic enzyme from Aster yellows witches'-broom (Candidatus Phytoplasma) (AYWB-ME) reveals a unique fold that differs from those of `canonical' MEs (Alvarez et al., p. 332). Here, residues of AYWB-ME involved in electrostatic interactions with NAD are shown. The AYWB-ME structure showcases a novel minimal structure design comprising a fully functional active site, making this enzyme an attractive starting point for rational genetic design.
research papers




Open  access
access
access
The expected log-likelihood gain can be used to predict the outcome of molecular replacement and optimize molecular-replacement strategies.

Open access
access
Newly improved tools for carbohydrate modelling in Coot are presented.
Measurement and modeling of the effects of dehydration and cryocooling on the conformational ensemble and solvent structure of monoclinic lysozyme crystals illuminate strong similarities in the local and global effects of these perturbations, which are attributed to their common effects on molecular packing within the crystal. These have an impact on the interpretation of differences between structures at T = 298 and 100 K and the choice of targets for variable-temperature crystallographic studies.
Open access
access
Maximum-likelihood rigid-body refinement can be carried out to improve oriented models before the translation-function step of molecular replacement.
Open access
access
ARCIMBOLDO_SHREDDER solves structures using fragments from low-homology models. Search fragments are improved through refinement or trimming against the experimental data. Consistent solutions are combined.


The type III pullulan hydrolase from Thermococcus kodakarensis (TK-PUL) possesses both pullulanase and α-amylase activities and has many potential applications in the industrial food-processing sector. Here, the crystal structure of TK-PUL is reported, which represents the first type III pullulan hydrolase structure to be solved, revealing N-terminal and C-terminal domains with significant differences from homologous structures.
PDB reference: type III pullulan hydrolase, 5ot1

The structure of the natural lipoaminopeptaibol helioferin is reported. It is a nonapeptide that contains only four proteinogenic amino-acid residues; two long aliphatic carbon chains protrude out from either side of the N-terminus of the helical peptide, giving rise to the corkscrew shape of the molecule.
PDB reference: helioferin, 6evh
Open access
access
Structures of native and variants (T84S and T84A) of M. tuberculosis class II fructose-1,6-bisphosphatase are presented and compared with those of other homologs. The structure is a 222-symmetric homotetramer. Citrate was bound at a dimer interface and was found to be an inhibitor.
The crystal structure of the malic enzyme from aster yellows witches'-broom (Candidatus Phytoplasma) is reported. The structure showcases a minimal scaffold harbouring malic enzyme activity, shedding light on the evolution of complex malic enzymes.
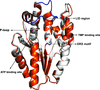
Conformational changes upon ligand binding and the path for communication between the substrates and the protein are important in understanding the catalytic mechanism of thymidylate kinase.
PDB references: T. thermophilus TMPK with Mg2+, 5xt8; with Cs+, 5xak; complex with TMP, 5x86; complex with ATP, 5x8a; complex with ATP and TMP, 5x8b; complex with AMPPCP and TMP, 5x8c; D90L mutant, 5x8d; K16M mutant, 5x8j; V158T mutant, 5x8k; Y92H mutant, 5x8v; Y162F mutant, 5x98; T18V mutant, 5x99; Y99F mutant, 5xal
Open access
access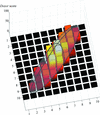
A method and software program, MeshBest, for the detection of individual crystals based on two-dimensional X-ray mesh scans are presented.
Open access
access
Multi-crystal serial crystallography data can be used for UV and X-ray radiation-damage-induced phasing.
addenda and errata
Free

Open access
access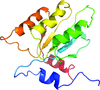
book reviews
Free








