

 journal menu
journal menu




issue contents
August 2020 issue
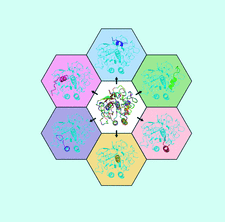
Cover illustration: Fragment generation and structure determination of proteinase K using ARCIMBOLDO_SHREDDER [Richards et al. (2020), Acta Cryst. D76, 703-712]. At the centre lies an overlay of all 759 fragments sequentially generated from the template model (PDB entry 4dzt). Examples of individual fragments derived from the model template are shown extracted out of the centre model in the context of the final structure of proteinase K.
editorial



Free 

Introducing the new Section Editor of Acta Cryst. D.
CCP4


Open access
access
Fragment-based determination of a proteinase K structure from MicroED data using ARCIMBOLDO_SHREDDER
A 1.6 Å resolution MicroED data set of proteinase K is phased using fragments derived from distantly related sequence homologues. ARCIMBOLDO_SHREDDER expands the phasing options for MicroED applications, overcoming the need for complete and highly accurate search models.
PDB reference: proteinase K, 6v8r
Open access
access
Two neural networks were trained to predict the correctness of protein residues by combining multiple validation metrics in Coot. Using the predicted correctness to automatically prune models led to significant improvements in the Buccaneer pipeline.
CCP-EM
Open access
access
Methods for obtaining the maximum out of a cryo-EM data-collection session are described.
ISDSB2019

Based on crystal structures of HIRAN in complex with duplex DNA, the mechanism of replication-fork regression underlying the sequence-independent recognition of nucleobases by HIRAN is discussed.
research papers
Open access
access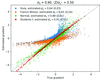
A quadrature is developed that allows the efficient evaluation of an intensity-based likelihood target function that includes experimental errors.

Download citation


Download citation

The benefits are shown of combining a high-density protein-crystallization technique (counter-diffusion) with in situ room-temperature data collection at synchrotron sources as an approximation to serial crystallography, without compromising data quality.

The X-ray crystallographic structure of an N-terminal fragment of group B streptococcus BibA (BibA126–398) and a low-resolution structure of the full-length N-terminal domain (BibA34–400) determined using small-angle X-ray scattering are described. The association of the N-terminal domain of BibA was localized to the C4BP α-chain.
PDB reference: BibA, 6poo
Open access
access
FragMAX is the fragment-screening platform of BioMAX, the macromolecular crystallography beamline of the MAX IV Laboratory. It provides support for users during sample preparation, data collection and data analysis through its in-house-developed software, FragMAXapp.
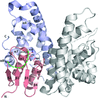
Crystal structures of XacGST, a glutathione S-transferase from the citrus canker pathogen Xanthomonas citri subsp. citri, with and without glutathione are reported.
Open access
access
A new procedure for experimental phasing at room temperature using diffraction data collected directly from the crystallization plates is described, demonstrated and tested.







