

 journal menu
journal menu





issue contents
January 2021 issue

Cover illustration: Using a partial atomic model from medium resolution cryo-EM to solve a large crystal structure: the portal protein of the T7 bacteriophage, gp8 [Fàbrega-Ferrer et al. (2021), Acta Cryst. D77, 11-18].
CCP4




Open  access
access
access
The employment of directed acyclic graphs to advance the tracking, control and appraisal of crystallographic phasing strategies is discussed.
Open access
access
A partial 30% atomic model from a 7.8 Å resolution cryo-EM volume was used to phase X-ray data by molecular replacement.


Open access
access
Virtual reality-specific tools for model building are possible, and can provide an order-of-magnitude speedup over mouse-and-keyboard tools in certain situations.
Open access
access
The LAHMA web server for structural analysis of homologous proteins is presented.
Open access
access
TEMPy2, an update of the TEMPy package to process, optimize and assess cryo-EM maps and the structures fitted to them, is presented.
Open access
access
Atomic models derived from cryo-EM data with map resolutions of better than 5 Å were automatically re-refined. The results of the computations are publicly available on a web page.
research papers
Open access
access
A motorized X–Y microscope stage is presented that combines human fine motor control with machine assistance and automated experiment documentation in order to transform productivity in protein crystal harvesting.

Open access
access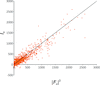
A likelihood-based method is improved to correct for dynamical scattering in weakly diffracting protein nanocrystals. The method has been tested on three protein crystals and yielded better refinement statistics and structure interpretations, including charge estimations of bound ions.
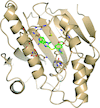
A high-resolution crystal structure of the Hsp90N–Debio0932 complex was successfully determined. Based on the crystal structure of the complex and on molecular-interaction analysis using thermal shift analysis and isothermal titration calorimetry, 30 new Debio0932 derivatives were designed and evaluated by molecular docking. Moreover, Debio0932 exhibited favourable in vitro anti-non-small-cell lung cancer activity.
PDB reference: Hsp90N in complex with Debio0932, 6lr9
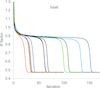
The solution of protein structure at atomic resolution using charge-flipping techniques operating on hundreds of virtual computers in the cloud is described. New techniques include the use of space-group symmetry restraints as well as the insertion of electron-density peaks.
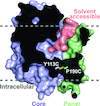
An engineered disulfide bridge traps and validates an outward-facing conformation in a bile acid transporter from Yersinia frederiksenii.
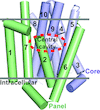
Crystal structures and docking of a bile acid transporter from Y. frederiksenii reveal details of the substrate-binding mechanism.
addenda and errata
Open access
access
book reviews
Free








