

 journal menu
journal menu research papers
research papers









The October 1998 release of the Cambridge Structural Database (1992) contains structural details (unit-cell dimensions and atom coordinates) for nearly 1300 distinct entries under space group P1 (No. 1); for 279 of these entries, the space-group designation is incorrect. The most common type of error, occurring for 157 entries with Z > 1, seems to have resulted from a simple misprint – the omission of the `overline' in the symbol 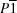 as it appears in the original publication; in these cases the reported coordinates, when applied to space group rather than P1, lead to reasonable intermolecular distances and to apparently reliable structures. In the remaining 123 cases the space group is incorrect for more fundamental reasons and the atom coordinates should be revised. In approximately one-third of the structures in which chiral molecules crystallize in P1 with Z = 2, the two molecules are related by an approximate center of inversion. In some cases this pseudocenter is surprisingly exact, with r.m.s. deviations from centrosymmetry as small as 0.1 Å, and may result in the same sort of refinement difficulties that inevitably arise when truly centrosymmetric structures are mistakenly refined in space group P1. It appears as though, for typical molecular compounds, standard crystal-structure techniques may be unable to distinguish between P1 and if the r.m.s. deviation from centrosymmetry is less than ∼0.1 Å.
as it appears in the original publication; in these cases the reported coordinates, when applied to space group rather than P1, lead to reasonable intermolecular distances and to apparently reliable structures. In the remaining 123 cases the space group is incorrect for more fundamental reasons and the atom coordinates should be revised. In approximately one-third of the structures in which chiral molecules crystallize in P1 with Z = 2, the two molecules are related by an approximate center of inversion. In some cases this pseudocenter is surprisingly exact, with r.m.s. deviations from centrosymmetry as small as 0.1 Å, and may result in the same sort of refinement difficulties that inevitably arise when truly centrosymmetric structures are mistakenly refined in space group P1. It appears as though, for typical molecular compounds, standard crystal-structure techniques may be unable to distinguish between P1 and if the r.m.s. deviation from centrosymmetry is less than ∼0.1 Å.
This article has 79 supporting items.



