journal menu
journal menu




issue contents
June 2000 issue

Cover illustration: Perspective view of the structure of the complex of 2,4,6-tris- (4-chlorophenoxy)-1,3,5-triazine with tribromobenzene, determined by neutron diffraction. Courtesy of J. A. K. Howard & C. K. Broder (University of Durham, UK), G. R. Desiraju, A. Nangia & R. K. R. Jetti (University of Hyderabad, India), C. C. Wilson & D. A. Keen (Rutherford Appleton Laboratory, Chilton, UK).
research papers



Download citation





Download citation

Reanalysis of the structure of the potassium ion conductor α-K3NdSi6O15·xH2O reveals it to be a hydrate. Structural features of its high-temperature polymorphs are described.
Download citation
Download citation

The new compound β-K3NdSi6O15 is a phyllosilicate with a silicate sheet topology that is distinct from that of the essentially isocompositional compound α-K3NdSi6O15·2H2O. The compactness of its structure results in a low ionic conductivity.

A model structure of Ag2SnO3 with a one-dimensional modulation has been elucidated by high-resolution electron microscopy, which is also supported by the `disorder' of the X-ray determined structure. The modulation occurs in 10–100 nm hexagonally twinned domains, which explains the apparent modulation in the three directions in electron diffraction patterns.
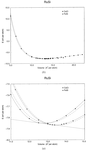
The relative stability of the two known polymorphs of RuSi, which have the ∊-FeSi and CsCl structures, has been investigated by first-principles pseudopotential calculations. It appears that RuSi having a CsCl-type structure will be the thermodynamically most stable phase for pressures greater than 3.6 GPa. The calculated bulk moduli are 202 and 244 GPa for the ∊-FeSi and CsCl phases, respectively.
Download citation
Download citation

A structural law based on the mutual arrangement of WO3-type layers is proposed to explain the symmetries and twins encountered in the series of monophosphate tungsten bronzes with pentagonal tunnels.
Download citation
Download citation

The structure of the hexagonal ν-Al80.61Cr10.71Fe8.68 [a = 40.68 (7), c = 12.546 (1) Å with 131 unique atoms] was solved by direct methods based on the structural model of a large icosahedral cluster derived from high-resolution electron microscopic images.
Download citation
Download citation
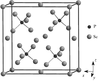
The crystal structures of β- and γ-Cu7PSe6, two of the three polymorphic forms of the argyrodite Cu7PSe6 compound, heptacopper phosphorus hexaselenide, are determined from single-crystal X-ray diffraction data. The copper-ion diffusion paths in the high-temperature form and the copper distribution in the second phase are studied by means of a Gram–Charlier non-harmonic development of the Debye–Waller factor. The analysis reveals a partial localization of copper, at the phase transition, in different low-coordination selenium sites, stressing its relative stability in such selenium environments.
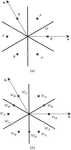
The structure of a new hexagonal perovskite phase, Sr9/8TiS3, was determined from single-crystal X-ray diffraction data, using the (3 + 1)-dimensional formalism; various original crenel functions were used to describe the S and Sr atoms accurately. A new TiS6 polyhedron was observed in the TiS3 chains, differentiating the hexagonal perovskite sulfides from the oxide counterparts.
Download citation
Download citation
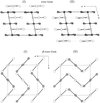
The crystal structures of five diammine-dihalide palladates are determined using an X-ray powder diffraction technique. The mechanism of the phase transition is discussed.
Download citation
Download citation

The structure of the tetragonal phase of the A-site substituted perovskite sodium bismuth titanate, Na0.5Bi0.5TiO3, is determined by neutron powder diffraction at 698 K. In-phase tilts accompanied by anti-parallel cation displacements parallel to the polar c axis, space group P4bm, are observed. This unusual combination results in a new variant of the perovskite structure.
Download citation
Download citation
The crystal structures of Bi24O31X10 (X = Cl, Br) have been redetermined; the previous structure model of simple construction has to be replaced by a more complicated one.
Download citation
Download citation

The crystal structures of (NH4)2[Cu(H2O)6](SO4)2 and of its form with isotopically substituted H218O are reported. They are very similar but differ in detail from the structure of the compound produced using deuterium oxide. This suggests that the influence of deuterium on the crystal structure is associated with the hydrogen-bonding interactions in the lattice rather than with its effect on the Jahn–Teller coupling through the increase in mass of the water ligand.
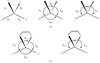
The geometries of four-coordinate CuI and CuII complexes in the Cambridge Structural Database have been analysed systematically and compared using symmetry-deformation coordinates and principal component analysis.

A methodology for the semi-automatic assignment and checking of metal oxidation states for metallo-organic complexes in the Cambridge Structural Database, which uses both chemical connectivity and bond-length data via ligand donor group templates and bond-valence sums, is described. This method has been used to validate the oxidation states in 743 four-coordinate copper complexes with a ca 99% success rate.
Algorithms for the automatic assignment of graph-set notation for intermolecular networks have been extended to molecules having internal crystallographic symmetry, for patterns up to the second level, and applied to a number of example molecules.
Download citation
Download citation
The crystal structure of six salts of terephthalic acid (three of which were determined ab initio using powder data) indicate that electrostatic interactions among the anions are important in determining the crystal packing.
Download citation
Download citation
Benzophenone was first identified as a polymorphic substance more than a century ago. In this paper the crystal structure of the metastable β-phase is presented for the first time and compared in detail with that of the well known stable phase.
Download citation
Download citation
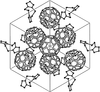
The crystal structure of (C60)8(C4S6N4)6 has been redetermined in the correct space group (I23) and reveals a novel type of extended, neutral, fullerene network. Tetrahedral holes are identified with a stoichiometry of five tetrahedral holes for eight C60 molecules.
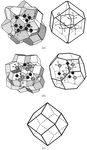
The calculation of molecular coordination numbers (MCNs) in the crystal structures of 23 067 organic compounds shows that Kitaigorodskii's close-packing model, assuming the predominance of MCN = 12, works correctly in only a few cases, whereas MCN = 14 is the most frequent. To explain this fact the close-packing model is extended with the model of the thinnest space covering by deformable molecules.
Download citation
Download citation
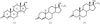
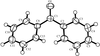
The crystal structures of six 2-oxa-steroids are analysed in terms of O—H⋯O and C—H⋯O hydrogen bonding. The first example of O—H⋯O/C—H⋯O interaction mimicry and binary solid solution is reported.

Cocrystallization of solvent molecules in the structures of organic and metalloorganic compounds has increased tremendously in the past 30 years. Relevant statistical data are presented, with special focus on structures that include two or more different types of solvent.

In crystal structure prediction it is usually assumed that only one molecule is present in the asymmetric unit. The number of possible crystal structures increases enormously if more than one independent molecule is allowed. The possibilities and limitations of theoretical prediction for such structures are investigated.
book reviews
Free