

 journal menu
journal menu




issue contents
April 2016 issue

Cover illustration: From data to knowledge using the Cambridge Structural Database [see Groom et al. (2016). Acta Cryst. B72, 171-179].
scientific commentaries



Free 
To scientists working with small-molecule or organometallic compounds, the Cambridge Structural Database constitutes an extremely important tool for reference to individual crystal structures and as a data source for statistical investigations. The article by Groom et al. provides updated information on the use, development and future of this database.
Free
The Source Function provides unique information about chemical bonding in the solid state, from theory as well as from experiment. It is now established that the concept of electronic delocalization in aromatic systems can be accurately studied using X-ray derived electron densities, and even more importantly the contributions are transferable between similar systems.
feature articles
Open access
access
This paper is the definitive article describing the creation, maintenance, information content and availability of the Cambridge Structural Database (CSD), the world's repository of small molecule crystal structures.

Free
The Source Function (SF), enabling the electron density to be viewed at a point as determined by source contributions from the various atoms or group of atoms of a system, is defined in terms of observables and it is amenable to experimental determination from accurate X-ray diffraction data. The use of the SF tool to discuss chemical bonding in molecules and crystals is reviewed, ahead of demonstrating that such a tool is also able to detect electron-delocalization effects and to assess their transferability properties in a series of paradigmatic molecular crystals, starting from knowledge of only the X-ray derived crystal electron densities.
research papers

Download citation



Download citation

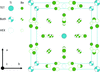
Incommensurately modulated borate structures of a new type are featured with ordering of oxygen atoms in nonlinear optical (NLO) materials Cs2TB4O9 (T = Ge, Si).
B-IncStrDB reference: 11442EEvkyY
Download citation
Download citation

γ-MoO3 nanobelts were prepared by hydrothermal synthesis. The novel polytype crystal structure and morphology of the belts were studied by synchrotron radiation powder diffraction, scanning electron microscopy, transmission electron microscopy and selected area electron diffraction. Their nm dimensions, in particular in two crystallographic directions, have a profound influence on electrochemical properties during cycling as the cathode material in Li-coin cells. The crystal structure for the γ-MoO3 nanobelts differs significantly from that of bulk α-MoO3. The question of whether the disorder structure type of γ-MoO3 nanobelts is the main reason for the increased charge capacity of MoO3 nanobelts from 200 for bulk materials up to 340 mAh g−1 for nanobelts remains open.
CCDC reference: 1444281
Download citation
Download citation

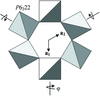
This paper describes the crystal structure of synthetic K2Sc[Si2O6]F in superspace group P42/mnm(α,α,0)000s(−α,α,0)0000 with modulation wavevectors q1 = 0.2982 (4)(a* + b*) and q2 = 0.2982 (4)(−a* + b*). The structure is a mixed octahedral–tetrahedral framework composed of [ScO4F2] octahedra, [Si4O12] rings and K in variable coordination.
B-IncStrDB reference: 11562ExhL04
Download citation
Download citation

The difficulty of obtaining crystals of a resolvable [RuL3]2+ cation is consistent with the remarkably complex structure of its PF6− salt.
CCDC reference: 1448676
Download citation
Download citation
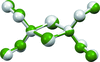
The crystal structure of diethylaminoalane, [H2Al—N(C2H5)2]2, was determined by X-ray powder diffraction, geometry optimization by density functional theory (DFT) and Raman spectroscopy. The DFT calculations were validated by calculating the ground state structures of two known aminoalanes while the Raman spectrum of diethylaminoalane was measured and compared to the simulated ones. Furthermore, the crystal structure of diethylaminoalane is compared with chemically and structurally similar compounds.
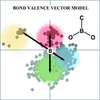
The bond-valence vector model is applied for the identification and analysis of subtle in- and out-of-plane distortions in three-coordinate boron in molecules containing a CBO2 skeleton.
Download citation
Download citation
The structural analysis of quartz across the α ↔ β transition is satisfactorily modelled with Landau theory. Cation–cation contractions give rise to negative-thermal expansion (NTE) in quartz and calcite.
Download citation
Download citation

Two new polymorphic forms of 5-nitrofurazone (5-nitro-2-furaldehyde semicarbazone) have been synthesized and structurally characterized by single-crystal and powder X-ray diffraction methods, vibrational spectroscopy, thermal and Hirshfeld surface analysis.
short communications

Structural complexity measured as the Shannon information per atom represents a negative contribution to the configurational entropy of a crystalline solid.
Download citation
Download citation
With the aid of atomic scale lattice dynamics simulations, the article resolves the long-standing controversy regarding the structure of TiBe12. The structure is confirmed to be tetragonal, whilst the hexagonal sub-cell used in recent publications is found to be an inappropriate model.
CCDC reference: 1455788
book reviews
Free

Free

Free

Free






