

 journal menu
journal menu




issue contents
August 2016 issue
Special issue on Crystal structure prediction
Guest Editors: Graeme M. Day and Carl Henrik Görbitz
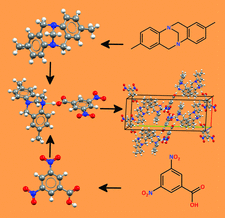
Cover illustration: Schematic of the crystal structure prediction of the multicomponent system of 3,5-dinitrobenzoic acid and Tröger's base, which was system (XXV) in the sixth blind test of crystal structure prediction methods. (XXV) was successfully predicted as a co-crystal in five submissions, starting only from the two-dimensional diagrams of the two components. While predicted as a co-crystal, low-temperature diffraction data suggest the proton is disordered between the two components [see Reilly et al. (2016). Acta Cryst. B72, 439-459].
editorial



Free 
The issue gives a flavour of progress and current research in the area of crystal structure prediction. Developments continue to be made in computational approaches for calculating the stabilities of molecular crystals, as well as for exploring crystal packing possibilities. As exemplified in the sixth blind test of crystal structure prediction, these methods are becoming applicable to complex target structures.
scientific commentaries
Free
This contribution comments on the advances of the latest Crystal Structure Prediction blind test and the challenges still lying ahead.
feature articles

Open access
access
The results of the sixth blind test of organic crystal structure prediction methods are presented and discussed, highlighting progress for salts, hydrates and bulky flexible molecules, as well as on-going challenges.
crystal structure prediction
A general and transferable force field for crystal structure predictions was developed.
Open access
access
An empirically parameterized intermolecular force field is developed for crystal structure modelling and prediction. The model is optimized for use with an atomic multipole description of electrostatic interactions.
Methods and results are presented for the crystal structure prediction of rigid molecules using force fields.
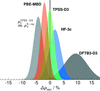
Route to high-accuracy crystal energy landscapes: From the sixth blind test challenge for crystal structure prediction we compile the POLY59 benchmark set and demonstrate the successful application of modern dispersion-corrected density functional approximations.
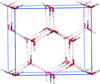
Molecular crystals expand at finite temperatures, which can have appreciable impacts on crystal structure and thermochemical properties. The ability to model thermal expansion using high-level electronic structure methods in the quasi-harmonic approximation and the importance of including thermal expansion effects is assessed for several small molecule crystals.
Open access
access
An investigation into using shape-similarity of molecules to generate putative crystal structures.


Exploring polymorphism of benzene and naphthalene with free energy based enhanced molecular dynamics
The crystal structures of benzene and naphthalene are successfully predicted for atmospheric and high-pressure conditions. Using enhanced molecular dynamics based sampling, we find that mixed structures, which typically cannot be discovered by standard crystal structure prediction methods, are prevalent in the solid forms of these compounds at high pressure.

Download citation



Download citation
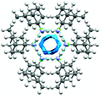
The crystal energy landscapes of two model pharmaceutically active chloride salts were used to rationalize the different hydration behaviour of the salts on the basis of the packing of the ions and calculated solvent-accessible volumes in the low-energy structures. The crystal structure of 1-(methylamino)adamantane was characterized and correctly predicted under blind test conditions as the second ranked structure in lattice energy, but the failure to predict the Z′ = 3 structure for the corresponding hydrochloride salt of this relatively simple base illustrates the limits of blind crystal structure prediction.

A putative crystal structure of tricyano-1,4-dithiino[c]-isothiazole with a layered packing motif is close in energy to the experimentally observed structure and is predicted to possess better electronic and optical properties.
research papers
Download citation
Download citation

Diffuse X-ray scattering from an organic salt has been analysed including modeling of the local structure, explaining relationships between symmetry and extinction patterns, and assessing the sensitivity of the calculated diffuse scattering to the model of the average structure.
Download citation
Download citation
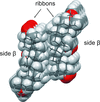
Molecular organization in unsolvated crystals of ester derivatives of glycyrrhetinic acid reproduces well two preferred modes of self-assembly in solvates of glycyrrhetinic acid and carbenoxolone.
Download citation
Download citation

Hirshfeld surface analysis, piezoelectricity and ferroelectricity are reported for the first time in L-histidinium dihydrogen arsenate orthoarsenic acid (LHAS) single crystals.
CCDC reference: 1478587
Open access
access
Bond-length distributions have been examined for 55 configurations of alkali-metal ions and 29 configurations of alkaline-earth-metal ions, for 4859 coordination polyhedra and 38 594 bond distances (alkali metals), and for 3038 coordination polyhedra and 24 487 bond distances (alkaline-earth metals).
A set of effective ionic radii corresponding to different coordination numbers (CNs) and compatible with the radii system by Shannon [Acta Cryst. (1976), A32, 751–767] has been derived for ammonium: 1.40 Å (CN = IV), 1.48 Å (CN = VI), 1.54 Å (CN = VIII) and 1.67 Å (CN = XII).
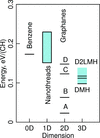
A new family of three-dimensional hydrocarbon polymers with no known structural analogs which are more energetically favorable than benzene is proposed.
Download citation
Download citation

Tetragonal InMo4O6 is a metal-rich compound with infinite chains of edge-sharing Mo6 clusters; the (3 + 1)-dimensional modulated structure hosts In6 and In7 oligomers in channels along [001] – this leads to two different kinds of disorder.
B-IncStrDB reference: 11852EKO6PR
CCDC reference: 1482621
Download citation
Download citation
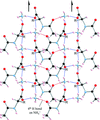
Structures of N-acetyl-DL-isoleucine, N-acetyl-DL-alloisoleucine and their ammonium salts, and the roles of ammonium ions in the crystal structure formation are reported.





