

 journal menu
journal menu





issue contents
May 2002 issue
Cover illustration: The different packing patterns of three crystal forms of S. typhi 3-dehydroquinase (p. 798).
research papers






The structure determination of 3-carboxy-cis,cis-muconate lactonizing enzyme from N. crassa by MAD at 3.0 Å is described and some of the difficulties in the structure determination and the methods used to address them are discussed. This represents one of the largest selenium substructures (80 Se atoms) solved.
PDB reference: CMLE, 1jof, r1jofsf

A new structure of concanavalin A, in a dimeric crystal form, has been refined using effective model-bias removal methods. The structure provides an opportunity to examine model accuracy and coordinate precision in high- or atomic resolution protein structures.
PDB reference: dimeric concanavalin A, 1gkb, r1gkbsf

The structure of binase, a member of the guanyl-specific microbial ribonuclease family of proteins, has been solved as a free enzyme at 1.65 Å resolution. This structure is compared with the binase complexes with 3′GMP and sulfate ions determined with data extending to 2.0 Å resolution.

The first biological crystal-growth experiment on the International Space Station used the Enhanced Gaseous Nitrogen Dewar and produced thaumatin crystals diffracting to 1.28 Å, which is comparable to previous space-grown thaumatin crystals and better than the best ground-control crystal (1.47 Å).

Real-space simulated-annealing refinement is implemented within an interactive molecular graphics package. This accelerates the rate-limiting step of model building and improves the starting point and final quality of subsequent conventional refinement.
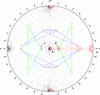
Ramachandran plots for all 20 amino acids were produced from 1042 protein subunits from the PDB, separately for those in SHEET, HELIX and Random coil. The classical Ramachandran plot needs to be revised in every detail. Most amino acids have two distinct conformations in the β-sheet region – one more frequent when the amino acids are really parts of SHEET and the other more typical for those occurring in Random coil. Amino acids in parallel and antiparallel β-strands have nearly the same conformations.

The crystal structures of the 18 kDa NII-domain fragment of duck ovotransferrin and its complex with nitrilotriacetate are reported at resolutions of 1.95 and 1.90 Å, respectively. The implication of these structures with respect to the mechanism of iron binding by the transferrins is discussed.
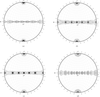
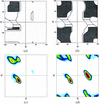
A combination of various approaches was used to determine precise twinning ratios. The importance of second-, third-, and fourth-order moments is discussed.
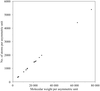
Formulae for the diffraction-component precision index are rearranged for easy calculation and to show how precision varies with such quantities as resolution and completeness of observation.
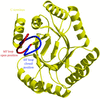
Crystal structures of two new crystal forms of the type I 3-dehydroquinate dehydratase (DHQase) have been determined and analysed.
PDB reference: type I DHQase, 1l9w, r1l9wsf

FIP is a French CRG beamline at the European Synchrotron Radiation Facility (ESRF) dedicated exclusively to crystallography of biological macromolecules, with a special emphasis on automated multiwavelength anomalous diffraction data collection in the 0.7–1.81 Å wavelength range.
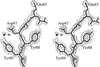
The three-dimensional structure of a human light-chain dimer (Sea) is reported to 1.94 Å resolution. This structure is likely to facilitate our understanding of why some seemingly harmless light chains display a propensity to aggregate as pathological amyloid fibrils.
PDB reference: immunoglobulin light-chain dimer, 1jvk, r1jvksf
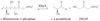
The C4-tetrameric enzyme crystallized with 20 protomers in the asymmetric unit. The atomic cstructure was solved using one heavy-atom derivative at 6 Å resolution and the molecular envelope of a related enzyme.
PDB reference: L-rhamnulose-1-phosphate aldolase, 1gt7, r1gt7sf
crystallization papers

The full-length (untruncated) protein dihydrolipoamide succinyltransferase, E2, of OGDC from pig heart, has been crystallized in space group I432, with unit-cell parameter a = 189.9 Å.

The crystallization of the complex between glycoprotein D (gD) from HSV-1 and the receptor HveA is decribed as well as the preparation of a selenomethionine-substituted gD in insect cells.
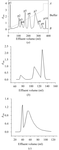
Single crystals of atratoxin have been grown from drops containing the necessary Cu2+ ions by the conventional hanging-drop vapour-diffusion method.

A novel haemoglobinase from pathogenic E. coli has been crystallized. The crystals diffract to 2.5 Å resolution.
Peptide deformylase from the pathogenic bacterium L. interrogans was identified. The enzyme was overexpressed in E. coli in high yield. The purified enzyme was crystallized and X-ray diffraction data were collected to 3 Å resolution.

Carbonic anhydrases catalyze the interconversion of carbon dioxide to bicarbonate. Human carbonic anhydrase isozyme III with a C-terminal hexahistidine tag was overexpressed in E. coli, purified and crystallized.
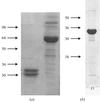
The C-terminal domain of catalase-peroxidase HPI (hydroperoxidase I) from E. coli has been crystallized.

The functional domain of human thrombopoietin was crystallized using a Fab fragment digested from its neutralizing antibody. Preliminary X-ray analysis suggests that each Fab fragment provides a suitable interface for crystallization.

The regulatory particle non-ATPase subunit, Nas6p, from S. cerevisiae has been crystallized by the hanging-drop method. A complete diffraction data set using synchrotron radiation was collected to 2.6 Å resolution.

Aspartate 1-decarboxylase (PanD) from H. pylori has been overexpressed and crystallized (space group I422; unit-cell parameters a = b = 81.83, c = 93.78 Å). Diffraction data have been collected to 1.55 Å using synchrotron X-rays.

UDP-N-acetylglucosamine acyltransferase (LpxA) from H. pylori has been overexpressed and crystallized (space group P6322; unit-cell parameters a = b = 90.69, c = 148.20 Å, α = β = 90, γ = 120°). Diffraction data have been collected to 2.1 Å using synchrotron X-rays.
The NAD+-dependent glycerol dehydrogenase from the extremely thermophilic bacterium T. maritima has been crystallized in the presence of glycerol by the hanging-drop vapour-diffusion method using 2-methyl-2,4-pentanediol (MPD) as the precipitating agent.

Crystals of recombinant hyperthermophilic archael histone HPhA were obtained by the hanging-drop vapour-diffusion method. Diffraction data have been collected to 2.3 Å.
short communications
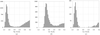
For sodium and potassium, geometry around the metal ion found in high-resolution protein structures in the Protein Data Bank is compared with that in appropriate small-molecule complexes in the Cambridge Structural Database. Target distances for refinement or for validation are proposed.

Exploiting genomic sequence databases for structural genomics target selection.
addenda and errata
Free 








