

 journal menu
journal menu





issue contents
February 2003 issue
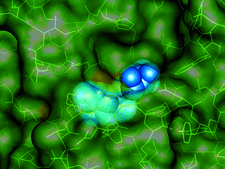
Cover illustration: A transparent representation of the active-site pocket of adenosine deaminase from bovine intestine. The inhibitor HDPR (blue spheres) is locked up by the protein lid around the pocket p. 299).
topical reviews





The physiological roles of lysosomal cysteine proteases now emerging from research are bringing them increasingly into focus as drug targets. A survey of the available crystal structures and a summary of their specificity determinants are presented here and some directions in inhibitor design are described.
research papers


The procedures used to solve the crystal structure of the SecA translocation ATPase are described. MIR phases to a limiting resolution of 4.4 Å were extended to the 2.7 Å resolution limit of the crystals using an iterative density modification and partial model building procedure that employed real-space cross-validation to minimize model bias.

A forensic analysis is presented of a trigonal crystal form with a complex pattern of twinning including merohedral twinning of the dominant lattice. The analysis indicates that twinning is caused by promiscuous parallel and antiparallel packing of helical protein fibers, permitted by the conformational flexibility of the protein combined with the inherent plasticity of the intermolecular interactions between the fibers.

High-quality three-dimensional crystals of membrane proteins can be produced using lipidic cubic phases. The detailed temperature-dependent phase behaviour of a wide range of monoolein–detergent mixtures is presented to aid the development of streamlined membrane-protein crystallization in cubo.
The crystal structure of a hybrid squash-type inhibitor in complex with porcine pancreatic elastase is described. The binding loop derived from the turkey ovomucoid inhibitor third domain adopts a `canonical' conformation upon elastase binding.
The crystal structure of piratoxin III may represent the T-state of this phospholipase A2 which, unusually, exhibits cooperative substrate binding.
PDB reference: piratoxin III, 1gmz, r1gmzsf

The crystal structure of the actin–DBP complex was determined at 2.4 Å resolution. The implications for the actin-sequestering role of DBP and the model of the actin-filament structure are discussed.
PDB reference: actin–DBP, 1ma9, r1ma9sf
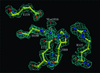
A method has been developed that automatically constructs a crystallographic refinement force field (topology and parameter files) for any molecule from a theoretical frequency calculation. The approach has been tested on five proteins containing metal sites or non-standard inhibitors or coenzymes and it is shown that the structures are improved in various aspects.

Use of multiple anomalous dispersion to phase highly merohedrally twinned crystals of interleukin-1β
The crystal structure of a double mutant (F42W/W120F) of interleukin-1β was determined by MAD using data from merohedrally twinned crystals. The detection and overcoming of twinning in interleukin-1β crystals and an analysis of the effect of the twinning on phasing by MAD is presented.
PDB reference: F42W/W120F interleukin-1β, 1l2h, r1l2hsf

The crystal structure of adenosine deaminase (ADA) from bovine intestine complexed with a transition-state analogue, 6-hydroxy-1,6-dihydropurine riboside (HDPR), was solved at 2.5 Å resolution. HDPR tightly interacts with ADA by means of six hydrogen bonds and is entirely enclosed within the active site.
PDB reference: ADA–HDPR complex, 1krm, r1krmsf
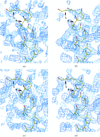
A new molecular-replacement code is introduced that balances local and global optimization through a global six-dimensional search of a low-resolution target function which is followed by multi-start local optimization.
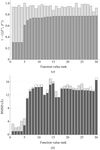
This paper discusses the results of an analysis of several data sets to determine the minimum amount of data needed to phase with SAD and MAD and the implications for the design of data-collection strategies seeking to minimize the total exposure time and dose absorbed by the crystal.
The crystal structure of bovine carboxypeptidase A has been refined at 1.25 Å resolution and allowed, together with theoretical simulations, an improved description of conformational disorder, solvent networks and protonation states in the resting enzyme.
PDB reference: bovine carboxypeptidase A at 1.25 Å, 1m4l, r1m4lsf
crystallization papers

Different crystal forms diffracting to high resolution have been obtained for two NADP(H)-dependent alcohol dehydrogenases: ScADHVI from S. cerevisiae and ADH8 from R. perezi.

An enzyme involved in cysteine biosynthesis in a hyperthermophilic archaeon has been crystallized. The crystal structure to be determined is expected to elucidate versatile substrate specificity in an ancestral-type enzyme.

The thermostable pectate lyase PL 47 from Bacillus sp. TS 47 was crystallized. A complete diffraction data set was collected to 1.8 Å resolution using synchrotron radiation.

Enzyme replacement therapy is clinically effective in reducing the morbidity associated with Gaucher's disease (a lysosomal storage disorder). A 2.7 Å data set has been collected from crystals of the responsible enzyme (β-glucocerebrosidase).
Cytochrome c peroxidase from P. stutzeri has been crystallized using sodium citrate and PEG 8000 as precipitants. The crystals belong to space group P21, with unit-cell parameters a = 69.29, b = 143.31, c = 76.83 Å, β = 100.78°.

The α-cyclodextrin glucanotransferase from B. macerans was purified homogeneously and crystallized. The crystals were suitable for X-ray analysis and diffracted to at least 2 Å.

The gene for E. coli ribonuclease P protein (also known as C5 protein) and its mutant C5-C113A have been expressed as GST-fusion proteins in E. coli at a high level. After cleavage of the fusion protein, highly purified functional C5 protein is obtained that can be crystallized and characterized by X-ray diffraction.
α-Methylacyl-CoA racemase from M. tuberculosis has been expressed in E. coli, purified and crystallized. Native crystals belonging to space group P6122 or P6522 diffract to 2.8 Å.

N-Acetyl-γ-glutamyl-phosphate reductase from T. thermophilus HB8 has been expressed in E. coli, purified and crystallized. The crystals belong to space group P6222 or P6422 and diffraction data have been collected to 2.2 Å resolution.

Crystals of the Y. pestis capsular antigen pre-assembly complex Caf1M–Caf1 have been obtained. Diffraction data from native, SeMet and Pt-labelled crystals were used to obtain a 2 Å electron-density map.
The S. rolfsii lectin was crystallized and X-ray diffraction data were collected to 1.1 Å resolution.

The boundaries of the Tudor domain of the spinal muscular neuron protein SMN have been defined using NMR and crystals that diffract to 1.8 Å resolution are reported.

A hybrid calpain containing 85% of μ-calpain sequence, and very close in properties to μ-calpain, has been crystallized. The structure may help in understanding the difference in Ca2+ requirement between μ- and m-calpain.

The 26 kDa TBP-interacting protein from the hyperthermophilic archaeon T. kodakaraensis KOD1 (Tk-TIP26) was crystallized and the crystals diffracted to 2.2 Å. Selenomethionine-substituted Tk-TIP26 was also crystallized and MAD data was collected. A Bijvoet difference Patterson map showed strong peaks sufficient to determine the positions of the Se atoms.

Two aldo-keto reductases from B. subtilis have been crystallized and their NADPH-dependent oxidoreductase activities have been kinetically characterized.

A fucose-specific lectin from A. aurantia was crystallized. 2.2 Å resolution data sets were collected and phased by the MAD method using an Hg derivative.

The dihydroorotase domain from hamster was crystallized by vapour diffusion. The crystals were found to exclusively consist of a disulfide-linked tetrameric form of the enzyme.

Preliminary results on the improved crystallization of manganese superoxide dismutase in the microgravity environment of the International Space Station are reported. Crystal volume was 80-times larger than the best earth crystal and diffraction to 1.26 Å resolution was observed.
short communications
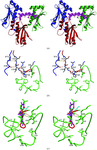
A crystal structure of the Y. enterocolitica molecular-chaperone protein SycE shows binding of its carboxy-terminal peptide to a hydrophobic patch on a neighboring molecule.
PDB reference: SycE, 1n5b, r1n5bsf
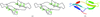
The unknown structure of the lima bean trypsin inhibitor was solved from the anomalous signal of the native sulfur using 3 Å in-house Cu Kα data and refined against 2 Å synchrotron data.
PDB reference: lima bean trypsin inhibitor, 1h34, r1h34sf
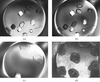
A modified microbatch technique is described which can be used to rapidly screen crystallization conditions. The characteristics of crystallization using silicone or paraffin oil are described.
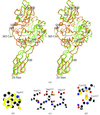
Homogeneous sample of chitinase A from Serratia marcescens yielded crystals that provided a structure to 1.55 Å resolution. Co-crystallization of the enzyme with its potent inhibitor allosamidin provided the structure of the complex to 1.9 Å resolution
addenda and errata
Free 






