

 journal menu
journal menu





issue contents
September 2006 issue
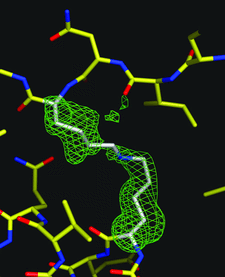
Cover illustration: Residues Lys66 and Lys778 form an intra-molecular cross-link in the copper-containing amine oxidase from Pichia pastoris. Omit electron density for the cross-link is visible in the structure refined at 1.23 Å resolution (p. 1073). The enzyme has apparently auto-catalytically oxidized one of its own lysine residues prior to the cross-linking.
research papers






A crystal form of influenza virus neuraminidase isolated from whale is reported in which calcium ions are absent, leading to structural heterogeneity of loops near the enzyme active site. Sulfate ions are observed in and near the active site.
PDB reference: influenza virus neuraminidase, 2b8h, r2b8hsf

The crystal structure of a bovine secretory signalling glycoprotein (SPC-40) has been determined at 2.1 Å resolution. The structure reveals new features of the sugar-binding groove and the presence of a novel flexible region implicated as being involved in receptor recognition.
PDB reference: SPC-40, 2esc, r2escsf

The crystal structure of AtETHE1 has been solved to a resolution of 1.48 Å. The structure has been analyzed and comparison is made to previously published structures of glyoxalase II enzymes.
PDB reference: AtETHE1, 2gcu, r2gcusf

The crystal structure of the N-terminal domain of human CEACAM1 is presented at 2.2 Å resolution. This structure is compared with its homologue from mouse and the binding surfaces are mapped for Neisseria Opa proteins and for other CEACAM molecules.
PDB reference: N-terminal domain of human CEACAM1, 2gk2, r2gk2sf
Open  access
access
access
Most Kunitz proteins like BPTI and α-dendrotoxin are stabilized by three disulfide bonds. The crystal structure shows how subtle repacking of non-covalent interactions may compensate for disulfide bond loss in a naturally occurring two-disulfide variant, conkunitzin-S1, the first discovered member of a new conotoxin family.
PDB reference: conkunitzin-S1, 1y62, r1y62sf
Open access
access
The diffraction properties of crystals of bovine F1-ATPase have been improved reproducibly by controlled dehydration, resulting in diffraction to 1.8 Å resolution.

The structure of a protein soaked in a bromate solution has been determined to a resolution of 1.25 Å.
PDB reference: lysozyme containing bromate ions, 2d6b, r2d6bsf
Open access
access
A new technique for the automated tracing of protein chains in experimental electron-density maps is described. The technique relies on the application of an electron-density likelihood target function to identify likely candidate Cα positions and to then grow these into connected protein chains.

Identifying non-crystallographic symmetry in protein electron-density maps: a feature-based approach
A robust, accurate and efficient feature-based algorithm to determine NCS operators in the initial stages of the structure determination process.

The crystal structure of the unique basic plastocyanin from A. variabilis was determined at 1.6 Å resolution.
PDB reference: plastocyanin, 2gim, r2gimsf
An experimental protocol is defined that provides a robust and reliable assessment of the radiation-sensitivity of protein crystals. Measurements on lysozyme crystals with varying heavy-ion content and on crystals of three other proteins suggest that radiation damage per dose is roughly constant at cryogenic temperatures.

An automated robotic system for protein streak-seeding is described which uses novel silicon microtools in place of the commonly used boar bristles.

Mg2+ induces a flexible state in helix α4 at the PhoB receiver domain α4-β5-α5 surface.
PDB reference: PhoB, 2yin, r2yinsf
Open access
access
An automated large-scale protein-crystallization and monitoring system for high-throughput protein structural analyses has been developed. Many protein crystals have been obtained using the system.

The method presented here evaluates protein crystallization states using classifiers.
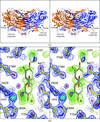
The high-resolution structure of P. pastoris lysyl oxidase reveals a lysine–lysine cross-link presumed to result from autocatalytic oxidation of a lysine residue of the enzyme.
PDB reference: PPLO, 1w7c, r1w7csf
The crystal structures of the apo form and a complex of M. jannaschii nucleoside kinase have been solved at 1.7 and 1.9 Å resolution, respectively. Several structural features typical for proteins from hyperthermophilic organisms, such as increased charged and a decreased hydrophobic accessible surface area and a higher fraction of charged residues, ionic networks and large aromatic clusters, were identified.
short communications
Open access
access
This paper describes a freely available software suite that allows the modelling of large conformational changes of protein structures for the interpretation of electron-microscopy reconstructions.
book reviews
Free

addenda and errata
Free








