

 journal menu
journal menu





issue contents
August 2006 issue
Get-Phases 2005
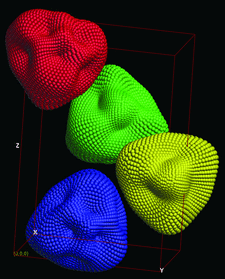
Cover illustration: This figure illustrates the packing of the small-angle X-ray scattering (SAXS) envelope in the crystallographic unit cell of nitrite reductase. The molecular replacement solution was found with the FSEARCH program (p. 909).




research papers

A large-scale, cost-effective, semi-automated platform for structural genomics has been set up at Peking University and this platform is aiming at a cost of US $10 000 per structure determination.

Beamline automation has significantly improved the throughput and accuracy of X-ray data collection from biological molecules. Further improvements will make the systems even more robust and offer a route to optimized and remote data collection.
Open  access
access
access
An expert system for semi-automatic or automatic analysis of X-ray diffraction data has been developed and used for over 140 new structure determinations. The typical end result is an interpretable electron-density map with a partially built structure.

Various criteria used for estimation of the anomalous signal in diffraction data are discussed.
A database is presented of heavy-atom derivatives that have been used in structure determination of membrane proteins. Compounds of organo-mercurials, PtII and trimethyl-lead are particularly successful.
Iterative dual-space fragment extension with protein SAD data is performed by a combination of the programs OASIS-2004, DM, RESOLVE (build only) and ARP/wARP. The procedure is beneficial to high-throughput protein structure determination.

Examples of Cr SAD structure solutions using a Cr/Cu dual-wavelength system in combination with a loopless free crystal-mounting method are shown.

Recent development on statistical direct phasing methods for heavy-atom substructure determination has been outlined.
Open access
access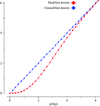
ACORN has been used to solve a number of protein structures and has been extended to work with data to resolution down to 1.7 Å.
This paper describes the development of the FSEARCH program for locating envelopes in the unit cell and possible ways to extend phases to crystallographic data resolution.
Open access
access
An automated ligand-fitting procedure has been developed and tested on 9327 ligands and (Fo − Fc)exp(iφc) difference density from macromolecular structures in the Protein Data Bank.
Open access
access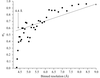
Methods and considerations for the structure solution and refinement of the ATPase p97/VCP at 4.7 to 3.5 Å resolution are described.

The crystal structures of bacterial ribonuclease III (RNase III) in complex with double-stranded (ds) RNA provide a structural basis for its non-catalytic and catalytic activities and insight into the mechanism of dsRNA processing by all members of the RNase III family.
The analysis of the translation apparatus has made it eminently clear that the complementarity of structural methods is needed to get an understandable and reasonably clear picture.







