

 journal menu
journal menu





issue contents
September 2007 issue
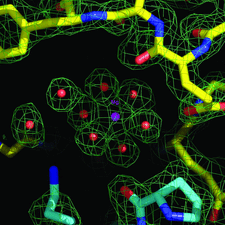
Cover illustration: The crystal structure of apo E. coli tryptophanase reveals a significant variation in the relative orientation of its two domains, reflecting functional flexibility (p. 969).
scientific comment




A small modification of the standard crystallographic cross-validation protocol (namely, the use of a dormant hold-out set of reflections) is described that makes it possible to separate optimization of the model parameterization and validation of the final model.
research papers

Structures deposited in the Protein Data Bank were analyzed for their quality using a combination of criteria. These analyses suggest that (i) the quality of protein crystal structures has remained the same over time, (ii) structural genomics generated structures are on average better and (iii) structures published in general science journals are on average significantly poorer in quality compared with the overall database.

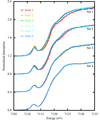
Single-crystal X-ray absorption spectroscopy (XAS) is used to show the temperature-dependence of X-ray photoreduction of the active site of putidaredoxin, an iron-containing metalloprotein, in a study that highlights the importance of using liquid-helium-based cooling during metalloprotein crystallography experiments and the utility of simultaneous XAS measurements.

Slit2 is a large multidomain protein containing an unusual domain organization of four tandem leucine-rich repeat (LRR) domains at its N-terminus. Here, the crystallization of the second and third LRR domains from human Slit2 and the structure of the third LRR domain are presented.
PDB reference: third LRR domain of human Slit2, 2v70, r2v70sf

The crystal structure of apo tryptophanase from E. coli (space group F222, unit-cell parameters a = 118.4, b = 120.1, c = 171.2 Å) was determined at 1.9 Å resolution using the molecular-replacement method and was refined to an R factor of 20.3% (Rfree = 23.2%).
PDB reference: apo tryptophanase, 2oqx, r2oqxsf

Here, the synthesis and crystal structures of the soluble extracellular domain of human neutral endopeptidase (NEP, residues 52–749) complexed with the NEP, competitive and potent dual NEP/DPP-IV inhibitor MCB3937 are described.
PDB reference: neprilysin–MCB3937 complex, 2qpj, r2qpjsf

The crystal structure of the extended-spectrum β-lactamase GES-1 has been solved to 1.1 Å resolution. Imipenem binding is analyzed by flexible-docking simulations into GES-1 and some point-mutant variants producing resistance to carbapenems.
PDB reference: GES-1, 2qpn, r2qpnsf

An unusual case of perfect pseudo-merohedral twinning in orthorhombic crystals of the enzyme Dicer is discussed.
PDB reference: Dicer, 2qvw, r2qvwsf

A comparison of hydrogenous and perdeuterated haloalkane dehalogenase shows similar overall structures with only slight alterations in surface regions. However, perdeuteration causes exclusion of a critical water nucleophile from the active site leading to a structure of an inactive, low pH enzyme form.
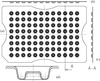
The development of a new type of vapour-diffusion crystallization plate is reported. It minimizes drop:precipitant volume to an ∼1:8 ratio and accelerates the rate of crystal growth.
short communications
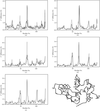
X-ray diffraction analysis of the dehydration-induced phase transition of monoclinic lysozyme crystals revealed that a sodium ion regulates the structure of the loop region Ser60–Asn74 and intermolecular contacts in the transformed crystal.
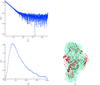
This paper discusses the execution of numerous simultaneous runs of the programs DAMMIN and GASBOR using Condor, which distributes them over a network of PCs; this has allowed a substantial acceleration of the processing of results. The application of Condor to determine the molecular shape of tissue transglutaminase is presented, thus demonstrating the efficiency of the approach.

Barium ions are used to provide phase estimates for xylanase II from T. longibrachiatum by SAD.







