

 journal menu
journal menu





issue contents
May 2008 issue
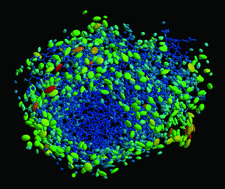
Cover illustration: 50% thermal ellipsoids for the structure of human aldose reductase at ultrahigh resolution 0.66 Å (p. 567). The water molecules are included and the colouring scheme is based on the equivalent atomic Beq factor. Lowest atomic Beq factors are shown in blue, while the highest are depicted in colours ranging from green to red.
research papers







The impact of hotspot mutation R282Q on the structure of human p53 DNA-binding domain has been characterized by X-ray crystallography and molecular-dynamics simulations.
PDB reference: p53 DNA-binding domain, 2pcx, r2pcxsf
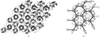
Low-resolution ab initio phased images of a six-Ig fragment from titin and their a posteriori comparison with the independently elucidated atomic model address the particular challenges that filamentous proteins pose to the low-resolution phasing methodology.
Open  access
access
access
Crystallization and analysis of the MIRAS heavy-atom structure solution of human cytochrome P450 46A1 using NaI and CsCl quick soaks.
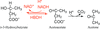
The crystal structures of D-3-hydroxybutyrate dehydrogenase from A. faecalis and of its complex with NAD+ and acetate suggest the mechanisms of substrate recognition and catalytic reaction. A structural comparison with other enzymes of the short-chain dehydrogenase/reductase family suggests that functional versatility has developed from a common ancestor protein.
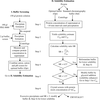
Increasing the solubility of proteins through buffer optimization and addition of glycerol to values above that found in a standard chromatography buffer led to a statistically significant improvement in crystallization screening results. Estimation of solubility in buffer provides a means for selection of an initial protein concentration for successful screening.
Open access
access
An OMIT procedure is presented that has the benefits of iterative model building density modification and refinement yet is essentially unbiased by the atomic model that is built.
Open access
access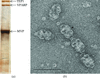
A vault from rat liver was crystallized in space group C2. Rotational symmetry searches indicated that the particle has 39-fold dihedral symmetry.
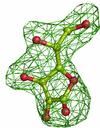
The crystal structure of monkey dimeric dihydrodiol dehydrogenase complexed with the inhibitor isoascorbic acid has been determined at 2.59 Å resolution.

The X-ray crystallographic structure of a dimer variant of fructose-1,6-bisphosphate aldolase demonstrates a stable oligomer that mirrors half of the native tetramer. The presence of product demonstrates that this is an active form.
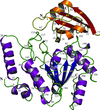
Uracil-DNA glycosylase from M. tuberculosis exhibits differences from the enzymes from other sources, particularly in the catalytic loops and the N- and C-terminal regions. The central region of the molecule is relatively invariant in sequence and structure among proteins from different organisms.
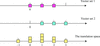
A new rotation function for molecular replacement by using both the self and cross Patterson vectors
A new rotation function has been designed for the molecular-replacement method in real space in which both the self and cross Patterson vectors are matched. This rotation function is shown to be sufficiently sensitive to detect a two-helix fragment in a myoglobin crystal structure.
A constrained refinement of the valence electron density against ultrahigh-resolution synchrotron X-ray diffraction data was performed using a multipolar atom model for the protein main-chain and side-chain atoms.
Structures of human saposin D are presented in two crystal forms at 1.3 and 2.0 Å. The protein adopts the compact monomeric saposin fold as seen in saposins A and C.
The first case of authentic structure determination by MAD phasing using high-resolution data from a (Ta6Br12)2+ derivative is analyzed to provide practical hints for the application of this useful phasing agent.
PDB reference: cytokinin-specific binding protein, 3c0v, r3c0vsf
short communications
Open access
access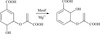
The structure of the menaquinone-specific isochorismate synthase (MenF) from Escherichia coli has been refined at a resolution of 2.0 Å in complex with magnesium. The magnesium-bound structure has a well defined and organized active site which better represents the active conformation of the enzyme than the currently available structure.







