

 journal menu
journal menu





issue contents
November 2008 issue
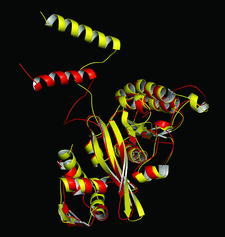
Cover illustration: A superposition of the native M. smegmatis RecA molecule (red) and the DNA-bound E. coli RecA molecule (yellow). Particularly striking is the movement of the helical N-terminal domain accompanied by a conformational change in the loop that connects the N-domain with the rest of the molecule (p. 1146).
research papers




PURY is database of geometric restraints derived from the crystal structures of small molecules deposited in the Cambridge Structural Database. The derived parameters cover a vast portion of chemical space and are ready for use in macromolecular crystal structure determination.


The ADP complex structure of the subunit B has helped in the understanding of the entry mode of the substrate in the A-ATP synthase and also the inhibitory mechanism of action of the antibiotic efrapeptin C.
PDB reference: A1A0 ATP synthase subunit B–ADP complex, 3dsr, r3dsrsf
Open  access
access
access
The Microcapillary Protein Crystallization System (MPCS) is a new protein-crystallization technology used to generate nanolitre-sized crystallization experiments for crystal screening and optimization. Using the MPCS, diffraction-ready crystals were grown in the plastic MPCS CrystalCard and were used to solve the structure of methionine-R-sulfoxide reductase.
PDB reference: methionine-R-sulfoxide reductase, 3cxk, r3cxksf
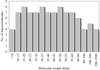
As part of a training set for automated image analysis, ∼150 000 images of crystallization experiments from 96 diverse macromolecules have been visually classified within seven categories. Outcomes and trends are analyzed.

As part of a training set for automated image analysis, crystallization screening experiments for 269 different macromolecules were visually analyzed and a set of crystal images extracted. Outcomes and trends are analyzed.
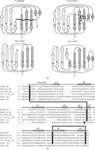
Crystal structures have been determined for two segments of the human F-spondin reeler domain at 1.45 and 2.70 Å resolution. The results provide insight into the structural plasticity of this domain.
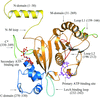
The structures of 19 crystals of mutants and nucleotide complexes of M. smegmatis RecA prepared under different conditions reported here, along with 11 previously reported structures, provide insights into the trajectory of allosteric transformations important in RecA function.
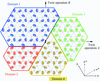
Equations are developed for determining the twin fractions from observed X-ray diffraction intensities in the case of tetartohedral twinning (i.e. where there are four differently oriented twin domains and each observed intensity contains contributions from four crystallographically independent reflections).
The crystal structure of the complex between LMPI-3 and F. oxysporum trypsin has been determined at 1.8 Å resolution. This study indicates that this small inhibitor interacts with the protease through the reactive site P3–P4′ and the P10–P6 residues.
PDB reference: LMPI-3–trypsin complex, 2vu8, r2vu8sf

The structure of the cancer-associated M. hyorhinis protein Mh-p37 has been determined to 1.9 Å resolution using a novel heavy-atom reagent, 5-amino-2,4,6-triiodoisophthalic acid, and SIRAS methods.
short communications
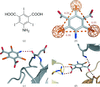
5-Amino-2,4,6-triiodoisophthalic acid is used as a phasing tool for protein structure determination. It is a representative of a novel class of compound for heavy-atom derivatization that combines heavy atoms with functional groups for binding to proteins.

This work presents a comparison of the crystal packing of three eukaryotic membrane proteins: human aquaporin 1, human aquaporin 5 and a spinach plasma membrane aquaporin. All were purified from expression constructs both with and without affinity tags.







