

 journal menu
journal menu





issue contents
April 2012 issue
Model building, refinement and validation
Proceedings of the CCP4 study weekend edited by Roberto A. Steiner, Bernhard Rupp and Charles Ballard

Cover illustration: Speakers at the 2011 CCP4 Study Weekend.




research papers
Open  access
access
access
Two developments in the process of automated protein model building in the Buccaneer software are described: the use of a database of protein fragments in improving the model completeness and the assembly of disconnected chain fragments into complete molecules.
Open access
access
ARCIMBOLDO combines the location of small fragments with Phaser and density modification with SHELXE of all possible Phaser solutions. Its uses are explained and illustrated through practical test cases.

Open access
access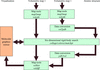
Recent developments of the Situs software suite for multi-scale modeling are reviewed. Typical workflows and conventions encountered during processing of biophysical data from electron microscopy, tomography or small-angle X-ray scattering are described.
Open access
access
phenix.refine is a program within the PHENIX package that supports crystallographic structure refinement against experimental data with a wide range of upper resolution limits using a large repertoire of model parameterizations. This paper presents an overview of the major phenix.refine features, with extensive literature references for readers interested in more detailed discussions of the methods.

Open access
access
Local structural similarity restraints (LSSR) provide a novel method for exploiting NCS or structural similarity to an external target structure. Two examples are given where BUSTER re-refinement of PDB entries with LSSR produces marked improvements, enabling further structural features to be modelled.
Open access
access
Recent developments in PHENIX are reported that allow the use of reference-model torsion restraints, secondary-structure hydrogen-bond restraints and Ramachandran restraints for improved macromolecular refinement in phenix.refine at low resolution.
Open access
access
DEN refinement and automated model building with AutoBuild were used to determine the structure of a putative succinyl-diaminopimelate desuccinylase from C. glutamicum. This difficult case of molecular-replacement phasing shows that the synergism between DEN refinement and AutoBuild outperforms standard refinement protocols.
PDB reference: succinyl-diaminopimelate desuccinylase, 3tx8
Open access
access
Low-resolution refinement tools implemented in REFMAC5 are described, including the use of external structural restraints, helical restraints and regularized anisotropic map sharpening.
Open access
access
A review of published tetartohedrally twinned macromolecular structures is presented, together with details of the recent structure determination of triclinic tetartohedrally twinned crystals of human complement factor I.
Open access
access
Coot is a molecular-graphics program designed to assist in the building of protein and other macromolecular models. The current state of ligand tools is presented.
Open access
access
The CCP4 template-restraint library defines restraints for biopolymers, their modifications and ligands that are used in macromolecular structure refinement. JLigand is a graphical editor for generating descriptions of new ligands and covalent linkages.
Open access
access
The challenges that arise in nucleic acid model building as a consequence of their simpler and more symmetric super-secondary structures are addressed.
Open access
access
Noncrystallographic symmetry is automatically detected and used to achieve higher completeness and greater accuracy of automatically built protein structures at resolutions of 2.3 Å or poorer.
Open access
access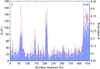
A likelihood-based metric for scoring the local agreement of a structure model with the observed electron density is described.
Open access
access
A simple rule of thumb based on resolution is not adequate to identify the best treatment of atomic displacements in macromolecular structural models. The choice to use isotropic B factors, anisotropic B factors, TLS models or some combination of the three should be validated through statistical analysis of the model refinement.
Open access
access
The implementation of a validation pipeline, based on community recommendations, for future depositions of X-ray crystal structures in the Protein Data Bank is described.
Open access
access
The decision-making algorithms and software used in PDB_REDO to re-refine and rebuild crystallographic protein structures in the PDB are presented and discussed.







