

 journal menu
journal menu





issue contents
August 2013 issue

Cover illustration: Top view of an ATP-independent Lon-like protease with open axial pores in tube representation with a semi-transparent surface (p. 1395). The N-terminal coil, the flexible Lon-insertion domain, the AAA-![[alpha]](/logos/entities/alpha_rmgif.gif) /
/![[beta]](/logos/entities/beta_rmgif.gif) , the AAA-, and the protease domains are colored in red, wheat, blue, cyan, and brown, respectively.
, the AAA-, and the protease domains are colored in red, wheat, blue, cyan, and brown, respectively.
feature articles




Free 
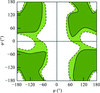
The most relevant aspects of fifty-year history of the Ramachandran plot are summarized, from the original ideas of Gopalasamudram Narayana Ramachandran to subsequent revisions and to applications in structural biology.
research papers

Open access
access
MAIN is interactive software designed to interactively perform the complex tasks of macromolecular crystal structure determination and validation. The features of MAIN and its tools for electron-density map calculations, model building, refinement in real and reciprocal space, and validation exploiting noncrystallographic symmetry in single and multiple crystal forms are presented.

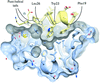
The design, validation, proof of mechanism and exploitation of a surface-entropy reduction mutant to solve the structure of the benchmark inhibitor Nutlin-3a bound to the cancer drug-discovery target MDM2 are reported.

Structures of the B. subtilis dUTPases in a novel complex with dU–PPi–Mg2+ and in complex with dUMP provide insights into the mechanism and identify features unique to the B. subtilis enzymes.
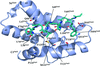
The crystal structure of the C-terminal peptide of secretin bound to the surface of its cognate pilotin reveals interactions that are essential for the assembly of a functional type II secretion system in D. dadantii and related secretion systems.
PDB reference: secretin–pilotin complex, 3uym

The crystal structure of HP1028, a protein from the human pathogen H. pylori, demonstrates that it belongs to the lipocalin family.
PDB reference: HP1028, 4inn

The crystal structure of an ATP-independent LonC protease revealed a proteolytic chamber with two open axial pores. A LonC-specific N-terminal coil tethers the AAA+ and protease domains together. Structures with covalent inhibitors bound to the proteolytic active sites provide mechanistic insights into the recognition of inhibitors and polypeptide substrates within the conserved Lon proteolytic chamber.
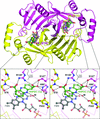
The structures of tricyclic intermediates trapped in crystals of PhzG suggest a basis for the chemistry and promiscuity of the final steps of phenazine biosynthesis.
The X-ray crystallographic structure of the disulfide-containing HCAII (dsHCAII) has been solved to 1.77 Å resolution and revealed that successful oxidation of the cysteine bond was achieved while also retaining desirable active-site geometry.
PDB reference: human carbonic anhydrase II, 4hba
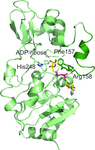
Crystal structures of human Sirt3 ligand complexes, complemented by biochemical analyses, reveal the binding site and mechanism for the small-molecule inhibitor SRT1720. The compound induces a unique co-factor binding-loop conformation and packs sandwich-like between a hydrophobic active-site patch and the nicotinamide moiety of the NAD+ cosubstrate.
The three-dimensional structure of an arylamine N-acetyltransferase enzyme from M. tuberculosis has been determined using cross-seeding with the homologous protein from M. marinum.
PDB reference: arylamine N-acetyltransferase, 4bgf
Details of five very high-resolution accurate structures of bovine trypsin are compared in the context of the reproducibility of models obtained from crystals grown under identical conditions.
PDB references: trypsin, model BT1, 4i8g; model BT2, 4i8h; model BT3, 4i8j; model BT4, 4i8k; model BT5, 4i8l
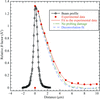
Reported here are measurements of the penetration depth and spatial distribution of photoelectron damage excited by 18.6 keV X-ray photons in a lysozyme crystal with a vertical submicrometre line-focus beam of 0.7 µm full-width half-maximum.
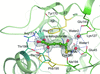
Structures of yeast Nit2 in complex with α-ketoglutarate and with oxaloacetate revealed the molecular-recognition mechanism of the enzyme for the first time, while that of the C169S mutant of yeast Nit2 showed a new ligand located in the catalytic cavity.
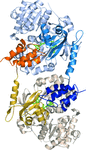
The structure of the functional interaction of NRPS adenylation and carrier protein domains, trapped with a mechanism-based inhibitor, is described. Crystals exhibit translational non-crystallographic symmetry, which challenged structure determination and refinement.
PDB reference: EntE-B, 4iz6

A novel three-chain nontoxic homologue of type II ribosome-inactivating proteins has been sequenced by mass spectrometry and its crystal structure has been determined. The evolutionary history of type II RIPs and the molecular basis for the loss of toxicity in some of them have been explored.
PDB reference: snake gourd seed lectin, 4hr6
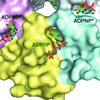
The ribonucleotide-binding properties of Hfq were probed using crystallography.
The first structural analysis of a procaspase-7 variant bound to a specific inhibitor, Ac-DEVD-CHO, revealed a structural asymmetry that the two L2 loops in homodimeric procaspase-7 served as inherent L2 and L2′ loops forming a complete active site in one monomer, which may be responsible to a basal activity level, but not in the other. This provides insight into the basal activity of procaspase-7 and the folding mechanism during caspase-7 activation.
The crystal structure of human PTPRQ and the following kinetic data provide the explanation for its dephosphosphorylating activity towards phosphatidyl inositides.
PDB reference: catalytic domain of PTPRQ, 4ikc
Structure refinement with nonspherical scattering factors from the invariom database is performed on thiostrepton, a heavily modified hexadecapeptide with antibiotic activity.
PDB reference: thiostrepton, 4hp2
Open access
access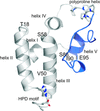
The crystal structure of the N-terminal part of T. thermophilus DnaJ unexpectedly showed an ordered GF domain and guided the design of a construct enabling the first structure determination of a complete DnaJ cochaperone molecule. By combining the crystal structures with spin-labelling EPR and cross-linking in solution, a dynamic view of this flexible molecule was developed.
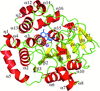
The enzymatic and structural characterization of the drug target purine-specific nucleosidase from the pathogen T. brucei provides a rationale for the tight-binding inhibition by iminoribitol-based compounds. The binding of a metalorganic inhibitor to nucleoside hydrolases is reported for the first time, together with the structural basis of metal-mediated noncompetitive inhibition.
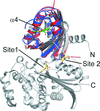
Structural studies of the protein encoded by orf12 of the clavulanic acid biosynthesis gene cluster from S. clavuligerus reveal a two-domain structure.

The crystal structure of lumazine synthase from C. glabrata complexed with its catalytic product 6,7-dimethyl-8-(D-ribityl)lumazine is reported.
PDB reference: lumazine synthase, 4kq6

The crystal structure of the SOCS2–elongin BC–Cul5(1–386) complex explains the specificity of Cul2 and Cul5 for elongin BC and their preferential association with Cul2 or Cul5 box-containing proteins.
PDB reference: SOCS2–elongin BC–Cul5N(RD), 4jgh
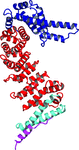
The crystal structure of full-length human CTNNBL1 reveals many unique structural features that establish it as a distinct member of the armadillo-repeat protein family.
Open access
access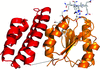
The first crystal structure of the vitamin B12-binding protein from a three-component O-demethylase enzyme system is reported. During O-demethylation methyl groups are transferred from phenyl methyl ethers to tetrahydrofolate via methyl-B12 intermediates.
PDB reference: CobDH, 4jgi
Open access
access
A systematic approach to the scaling and merging of data from multiple crystals in macromolecular crystallography is introduced and explained.







