Hindered rotation about the partial double C—N bonds between the amine and pyridine moieties in the title molecule, C
16H
14N
4, results in two different conformations of the
N-aryl-2-aminopyridine units. One, assuming an
E conformation, is involved in a pair of N—H

N hydrogen bonds that generate a centrosymmetric
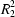
(8) motif. The second, adopting a
Z conformation, is not engaged in any hydrogen bonding and is flattened, the dihedral angle between the benzene and pyridine rings being 12.07 (7)°. This conformation is stabilized by an intramolecular C—H
N interaction [C
N = 2.9126 (19) Å, H
N = 2.31 Å and C—H
N = 120°].
Supporting information
CCDC reference: 235352
1,2-Diaminobenzene (2.7 g, 25 mmol) was dissolved in 2-chloropyridine (7 ml, 75 mmol) and refluxed for 1 h. After cooling, the precipitated hydrochloride was filtered off and washed with diethyl ether. The crude hydrochloride (8.5 g) was dissolved in water, 25% aqueous ammonia (25 ml) was added and the precipitated product was extracted with ethyl acetate. The organic layer was dried (Na2SO4), evaporated to dryness and the residue was crystallized from ethanol [yield 4.59 g, 70%; m.p. 440–441 K, literature 439–440 K (Fischer, 1902)]; 1H NMR (CDCl3): δ 8.16 (m, 2 H), 7.71 (dd, J = 3.6 and 6.0 Hz, 2 H), 7.42 (m, 2 H), 7.12 (dd, J = 3.5 and 6.0 Hz, 2 H), 6.86 (br s, 2 H), 6.70 (m, 2 H) and 6.59 (m, 2 H); 13C NMR (DMSO-d6): δ 156.8, 147.8, 137.8, 133.7, 123.7, 114.5 and 109.6.
H atoms attached to N atoms were located in a difference Fourier map and refined isotropically. H atoms attached to C atoms were placed in calculated positions (0.96 Å) and treated as riding. Their isotropic displacement parameters were refined.
Data collection: CrysAlis (Oxford Diffraction, 2000); cell refinement: CrysAlis; data reduction: CrysAlis; program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: 'Stereochemical Workstation (Siemens, 1989)'; software used to prepare material for publication: SHELXL97.
N,
N'-Bis(2-pyridyl)-1,2-diaminobenzene
top
Crystal data top
| C16H14N4 | F(000) = 552 |
| Mr = 262.31 | Dx = 1.334 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P2yn | Cell parameters from 4833 reflections |
| a = 9.1835 (6) Å | θ = 4–25° |
| b = 7.8999 (6) Å | µ = 0.08 mm−1 |
| c = 18.2023 (13) Å | T = 110 K |
| β = 98.598 (6)° | Block, colorless |
| V = 1305.71 (16) Å3 | 0.40 × 0.40 × 0.20 mm |
| Z = 4 | |
Data collection top
Kuma KM-4 CCD
diffractometer | 2319 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.031 |
| Graphite monochromator | θmax = 26.4°, θmin = 3.5° |
| ω scans | h = −11→11 |
| 6639 measured reflections | k = −9→9 |
| 2655 independent reflections | l = −22→17 |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.046 | Hydrogen site location: mixed |
| wR(F2) = 0.125 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.08 | w = 1/[σ2(Fo2) + (0.0717P)2 + 0.3063P]
where P = (Fo2 + 2Fc2)/3 |
| 2655 reflections | (Δ/σ)max < 0.001 |
| 201 parameters | Δρmax = 0.20 e Å−3 |
| 0 restraints | Δρmin = −0.25 e Å−3 |
Crystal data top
| C16H14N4 | V = 1305.71 (16) Å3 |
| Mr = 262.31 | Z = 4 |
| Monoclinic, P21/n | Mo Kα radiation |
| a = 9.1835 (6) Å | µ = 0.08 mm−1 |
| b = 7.8999 (6) Å | T = 110 K |
| c = 18.2023 (13) Å | 0.40 × 0.40 × 0.20 mm |
| β = 98.598 (6)° | |
Data collection top
Kuma KM-4 CCD
diffractometer | 2319 reflections with I > 2σ(I) |
| 6639 measured reflections | Rint = 0.031 |
| 2655 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.046 | 0 restraints |
| wR(F2) = 0.125 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.08 | Δρmax = 0.20 e Å−3 |
| 2655 reflections | Δρmin = −0.25 e Å−3 |
| 201 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| N1 | 0.06342 (13) | 0.69506 (15) | 0.30881 (6) | 0.0239 (3) | |
| C2 | 0.10734 (15) | 0.77340 (17) | 0.25114 (7) | 0.0211 (3) | |
| C3 | 0.25440 (17) | 0.82451 (19) | 0.25111 (9) | 0.0284 (3) | |
| H3A | 0.2826 | 0.8810 | 0.2088 | 0.029 (4)* | |
| C4 | 0.35691 (16) | 0.79234 (19) | 0.31243 (9) | 0.0299 (3) | |
| H4A | 0.4574 | 0.8273 | 0.3137 | 0.033 (4)* | |
| C5 | 0.31310 (16) | 0.70857 (19) | 0.37287 (8) | 0.0288 (3) | |
| H5A | 0.3824 | 0.6821 | 0.4162 | 0.036 (5)* | |
| C6 | 0.16702 (17) | 0.66474 (19) | 0.36800 (8) | 0.0278 (3) | |
| H6A | 0.1364 | 0.6087 | 0.4099 | 0.033 (4)* | |
| N7 | 0.00640 (13) | 0.80816 (15) | 0.18831 (6) | 0.0237 (3) | |
| H7 | 0.0430 (18) | 0.874 (2) | 0.1560 (9) | 0.026 (4)* | |
| C8 | −0.14579 (15) | 0.77847 (16) | 0.17315 (7) | 0.0207 (3) | |
| C9 | −0.22453 (15) | 0.85996 (17) | 0.11013 (7) | 0.0211 (3) | |
| C10 | −0.37580 (15) | 0.83763 (17) | 0.09312 (8) | 0.0242 (3) | |
| H10A | −0.4284 | 0.8914 | 0.0499 | 0.028 (4)* | |
| C11 | −0.45232 (16) | 0.73926 (18) | 0.13750 (8) | 0.0263 (3) | |
| H11A | −0.5575 | 0.7288 | 0.1265 | 0.030 (4)* | |
| C12 | −0.37415 (16) | 0.65587 (18) | 0.19818 (8) | 0.0270 (3) | |
| H12A | −0.4260 | 0.5857 | 0.2286 | 0.029 (4)* | |
| C13 | −0.22250 (16) | 0.67226 (18) | 0.21539 (8) | 0.0249 (3) | |
| H13A | −0.1700 | 0.6107 | 0.2565 | 0.029 (4)* | |
| N14 | −0.14696 (13) | 0.96227 (15) | 0.06474 (6) | 0.0235 (3) | |
| H14 | −0.069 (2) | 0.919 (2) | 0.0446 (10) | 0.041 (5)* | |
| C15 | −0.18829 (14) | 1.12400 (17) | 0.04170 (7) | 0.0196 (3) | |
| N16 | −0.11729 (12) | 1.18788 (14) | −0.01178 (6) | 0.0212 (3) | |
| C17 | −0.14821 (16) | 1.34886 (18) | −0.03364 (8) | 0.0251 (3) | |
| H17A | −0.0969 | 1.3958 | −0.0711 | 0.029 (4)* | |
| C18 | −0.24899 (17) | 1.45022 (18) | −0.00539 (8) | 0.0292 (3) | |
| H18A | −0.2680 | 1.5639 | −0.0228 | 0.032 (4)* | |
| C19 | −0.32205 (17) | 1.38154 (19) | 0.04925 (8) | 0.0285 (3) | |
| H19A | −0.3934 | 1.4480 | 0.0700 | 0.033 (4)* | |
| C20 | −0.29234 (15) | 1.21857 (18) | 0.07360 (8) | 0.0238 (3) | |
| H20A | −0.3416 | 1.1706 | 0.1116 | 0.030 (4)* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| N1 | 0.0241 (6) | 0.0269 (6) | 0.0211 (6) | 0.0017 (5) | 0.0050 (5) | 0.0007 (5) |
| C2 | 0.0215 (7) | 0.0205 (6) | 0.0222 (6) | 0.0030 (5) | 0.0057 (5) | −0.0012 (5) |
| C3 | 0.0248 (7) | 0.0313 (8) | 0.0301 (7) | −0.0001 (6) | 0.0079 (6) | 0.0060 (6) |
| C4 | 0.0217 (7) | 0.0322 (8) | 0.0357 (8) | 0.0010 (6) | 0.0043 (6) | −0.0025 (6) |
| C5 | 0.0267 (8) | 0.0334 (8) | 0.0249 (7) | 0.0078 (6) | −0.0002 (6) | −0.0023 (6) |
| C6 | 0.0318 (8) | 0.0302 (8) | 0.0218 (7) | 0.0039 (6) | 0.0057 (6) | 0.0016 (6) |
| N7 | 0.0211 (6) | 0.0291 (6) | 0.0220 (6) | −0.0003 (5) | 0.0064 (5) | 0.0061 (5) |
| C8 | 0.0219 (7) | 0.0204 (6) | 0.0204 (6) | 0.0006 (5) | 0.0053 (5) | −0.0025 (5) |
| C9 | 0.0238 (7) | 0.0196 (6) | 0.0215 (7) | 0.0035 (5) | 0.0084 (5) | −0.0003 (5) |
| C10 | 0.0237 (7) | 0.0240 (7) | 0.0251 (7) | 0.0049 (5) | 0.0043 (5) | −0.0015 (5) |
| C11 | 0.0212 (7) | 0.0252 (7) | 0.0330 (8) | −0.0017 (5) | 0.0056 (6) | −0.0037 (6) |
| C12 | 0.0283 (8) | 0.0254 (7) | 0.0290 (7) | −0.0053 (6) | 0.0104 (6) | −0.0006 (6) |
| C13 | 0.0276 (7) | 0.0250 (7) | 0.0225 (7) | −0.0012 (6) | 0.0052 (6) | 0.0007 (5) |
| N14 | 0.0227 (6) | 0.0243 (6) | 0.0257 (6) | 0.0071 (5) | 0.0111 (5) | 0.0057 (5) |
| C15 | 0.0176 (6) | 0.0225 (7) | 0.0182 (6) | 0.0001 (5) | 0.0011 (5) | 0.0001 (5) |
| N16 | 0.0193 (6) | 0.0233 (6) | 0.0215 (6) | 0.0012 (4) | 0.0043 (4) | 0.0019 (4) |
| C17 | 0.0256 (7) | 0.0243 (7) | 0.0256 (7) | −0.0004 (5) | 0.0045 (6) | 0.0045 (5) |
| C18 | 0.0333 (8) | 0.0196 (7) | 0.0353 (8) | 0.0044 (6) | 0.0069 (6) | 0.0028 (6) |
| C19 | 0.0304 (8) | 0.0250 (7) | 0.0315 (8) | 0.0064 (6) | 0.0092 (6) | −0.0025 (6) |
| C20 | 0.0233 (7) | 0.0266 (7) | 0.0225 (7) | 0.0024 (5) | 0.0070 (5) | −0.0008 (5) |
Geometric parameters (Å, º) top
| N1—C2 | 1.3320 (18) | C10—H10A | 0.96 |
| N1—C6 | 1.3480 (19) | C11—C12 | 1.390 (2) |
| C2—N7 | 1.3877 (18) | C11—H11A | 0.96 |
| C2—C3 | 1.410 (2) | C12—C13 | 1.387 (2) |
| C3—C4 | 1.372 (2) | C12—H12A | 0.96 |
| C3—H3A | 0.96 | C13—H13A | 0.96 |
| C4—C5 | 1.394 (2) | N14—C15 | 1.3802 (17) |
| C4—H4A | 0.96 | N14—H14 | 0.92 (2) |
| C5—C6 | 1.375 (2) | C15—N16 | 1.3479 (17) |
| C5—H5A | 0.96 | C15—C20 | 1.4051 (19) |
| C6—H6A | 0.96 | N16—C17 | 1.3500 (18) |
| N7—C8 | 1.4033 (18) | C17—C18 | 1.380 (2) |
| N7—H7 | 0.889 (17) | C17—H17A | 0.96 |
| C8—C13 | 1.3975 (19) | C18—C19 | 1.390 (2) |
| C8—C9 | 1.4160 (19) | C18—H18A | 0.96 |
| C9—C10 | 1.3886 (19) | C19—C20 | 1.376 (2) |
| C9—N14 | 1.4211 (17) | C19—H19A | 0.9600 |
| C10—C11 | 1.386 (2) | C20—H20A | 0.96 |
| | | |
| C2—N1—C6 | 116.86 (12) | C10—C11—C12 | 118.84 (13) |
| N1—C2—N7 | 119.86 (12) | C10—C11—H11A | 120.6 |
| N1—C2—C3 | 122.43 (13) | C12—C11—H11A | 120.6 |
| N7—C2—C3 | 117.71 (13) | C13—C12—C11 | 121.13 (13) |
| C4—C3—C2 | 119.18 (14) | C13—C12—H12A | 119.5 |
| C4—C3—H3A | 120.3 | C11—C12—H12A | 119.4 |
| C2—C3—H3A | 120.5 | C12—C13—C8 | 120.23 (13) |
| C3—C4—C5 | 119.09 (14) | C12—C13—H13A | 119.9 |
| C3—C4—H4A | 120.5 | C8—C13—H13A | 119.8 |
| C5—C4—H4A | 120.4 | C15—N14—C9 | 124.34 (11) |
| C6—C5—C4 | 117.55 (13) | C15—N14—H14 | 114.8 (12) |
| C6—C5—H5A | 121.2 | C9—N14—H14 | 120.6 (12) |
| C4—C5—H5A | 121.2 | N16—C15—N14 | 115.17 (11) |
| N1—C6—C5 | 124.88 (14) | N16—C15—C20 | 122.12 (12) |
| N1—C6—H6A | 117.5 | N14—C15—C20 | 122.67 (12) |
| C5—C6—H6A | 117.6 | C15—N16—C17 | 117.54 (12) |
| C2—N7—C8 | 130.68 (12) | N16—C17—C18 | 124.05 (13) |
| C2—N7—H7 | 112.9 (11) | N16—C17—H17A | 118.0 |
| C8—N7—H7 | 115.6 (11) | C18—C17—H17A | 118.0 |
| C13—C8—N7 | 124.15 (12) | C17—C18—C19 | 117.54 (13) |
| C13—C8—C9 | 118.70 (12) | C17—C18—H18A | 121.3 |
| N7—C8—C9 | 117.14 (12) | C19—C18—H18A | 121.2 |
| C10—C9—C8 | 119.71 (12) | C20—C19—C18 | 120.14 (13) |
| C10—C9—N14 | 120.96 (12) | C20—C19—H19A | 119.9 |
| C8—C9—N14 | 119.33 (12) | C18—C19—H19A | 119.9 |
| C11—C10—C9 | 121.26 (13) | C19—C20—C15 | 118.61 (13) |
| C11—C10—H10A | 119.4 | C19—C20—H20A | 120.7 |
| C9—C10—H10A | 119.4 | C15—C20—H20A | 120.7 |
| | | |
| N1—C2—N7—C8 | 2.0 (2) | C10—C9—N14—C15 | 49.39 (19) |
| C3—C2—N7—C8 | −177.63 (13) | C8—C9—N14—C15 | −131.25 (14) |
| C2—N7—C8—C9 | 167.00 (13) | C9—N14—C15—N16 | −168.21 (12) |
| C2—N7—C8—C13 | −14.1 (2) | C9—N14—C15—C20 | 14.3 (2) |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C13—H13A···N1 | 0.96 | 2.31 | 2.9126 (19) | 120 |
| N14—H14···N16i | 0.92 (2) | 2.07 (2) | 2.9874 (16) | 173.2 (16) |
| Symmetry code: (i) −x, −y+2, −z. |
Experimental details
| Crystal data |
| Chemical formula | C16H14N4 |
| Mr | 262.31 |
| Crystal system, space group | Monoclinic, P21/n |
| Temperature (K) | 110 |
| a, b, c (Å) | 9.1835 (6), 7.8999 (6), 18.2023 (13) |
| β (°) | 98.598 (6) |
| V (Å3) | 1305.71 (16) |
| Z | 4 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.08 |
| Crystal size (mm) | 0.40 × 0.40 × 0.20 |
| |
| Data collection |
| Diffractometer | Kuma KM-4 CCD
diffractometer |
| Absorption correction | – |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 6639, 2655, 2319 |
| Rint | 0.031 |
| (sin θ/λ)max (Å−1) | 0.625 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.046, 0.125, 1.08 |
| No. of reflections | 2655 |
| No. of parameters | 201 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.20, −0.25 |
Selected geometric parameters (Å, º) top| N1—C2 | 1.3320 (18) | C9—N14 | 1.4211 (17) |
| N1—C6 | 1.3480 (19) | N14—C15 | 1.3802 (17) |
| C2—N7 | 1.3877 (18) | C15—N16 | 1.3479 (17) |
| N7—C8 | 1.4033 (18) | N16—C17 | 1.3500 (18) |
| | | |
| C2—N1—C6 | 116.86 (12) | C15—N14—C9 | 124.34 (11) |
| C2—N7—C8 | 130.68 (12) | C15—N16—C17 | 117.54 (12) |
| | | |
| N1—C2—N7—C8 | 2.0 (2) | C10—C9—N14—C15 | 49.39 (19) |
| C2—N7—C8—C13 | −14.1 (2) | C9—N14—C15—C20 | 14.3 (2) |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C13—H13A···N1 | 0.96 | 2.31 | 2.9126 (19) | 120.1 |
| N14—H14···N16i | 0.92 (2) | 2.07 (2) | 2.9874 (16) | 173.2 (16) |
| Symmetry code: (i) −x, −y+2, −z. |


 journal menu
journal menu organic compounds
organic compounds











![[Figure 1]](https://journals.iucr.org/c/issues/2004/03/00/av1160/av1160fig1thm.gif)




N,N'-Bis(2-pyridyl)aryldiamines are versatile building blocks for syntheses of extended supramolecular arrays (Bensemann et al., 2002; Gdaniec et al., 2002; Bensemann et al., 2003). Bearing two self-complementary 2-aminopyridine units, these molecules are able to form in crystals one-dimensional hydrogen-bonding networks using the cyclic R22(8) motif, or to assemble via a catemeric C(4) motif into one-, two- or even three-dimensional networks (Bensemann et al., 2002). In the first case, the N-aryl-2-aminopyridine units have to assume the E conformation, whereas the C(4) motif requires the units to adopt the Z conformation. A search of Cambridge Structural Database (CSD; Version 5.24, 272066 entries, plus three 2003 updates; Allen, 2002) revealed that the mean values of the dihedral angles between pyridine and aryl planes in the N-aryl-2-aminopyridine moieties, which are involved in hydrogen bonding as H-atom donor and H-atom acceptor, are similar for the E and Z forms [51 (7) and 47 (3)°, respectively].
However, in contrast to the E form, which cannot be planar for steric reason, the N-aryl-2-aminopyridine unit in the Z form is also able to adopt a conformation with nearly coplanar pyridine and aryl rings. In this arrangement, the pyridine N atom, being a hydrogen-bond acceptor, is blocked by the intramolecular C—H···N interaction and thus the N-aryl-2-aminopyridine unit can act only as an NH donor in the intermolecular hydrogen bonding. Among symmetrical N,N'-bis(2-pyridyl)aryldiamines studied so far, E,E or Z,Z conformations have been observed, but the E,Z form has never been reported (Bensemann et al., 2002; Kempe & Hillebrand, 2000). We report here the crystal structure of N,N'-bis(2-pyridyl)-1,2-diaminobenzene, (I), the first example of this class of compounds that exists in the E,Z form in the crystalline state.
The structure of (I), with the atom numbering scheme, is presented in Fig. 1, which shows one of the N-aryl-2-aminopyridine units (N1/C13) adopting a strongly flattened Z conformation, with N1—C2—N7—C8 and C2—N7—C8—C13 torsion angles equal to 2.0 (2) and −14.1 (2)°, respectively. This conformation is stabilized by the intramolecular C—H···N interaction between pyridine atom N1 and benzene atom H13 [C13···N1 = 2.9126 (19) Å, H13···N1 = 2.31 Å and C13—H13···N1 = 120°]. The second N-aryl-2-aminopyridine unit (C8–C20), adopting the E conformation, is significantly non-planar, with the aromatic rings strongly twisted about the C—N bonds to the amine group [C9—N14—C15—C20 = 14.3 (2)° and C10—C9—N14—C15 = 49.39 (19)°]. In this conformation, the H atoms of the two N—H groups in the molecule of (I) are separated by 2.16 (2) Å. A search of the CSD for N-aryl-2-aminopyridines showed that the dihedral angle between the pyridine ring and the plane of the amine group is generally smaller than that between the amine and N-aryl groups. This difference is probably a consequence of a stronger conjugation of the amine N π electrons with the pyridine π system. It? is further confirmed by the shortening of the C—N bond to the amine N atom; the mean value of the C—N bond length to the pyridine ring is significantly shorter than that to the aryl substituent [1.369 (7) and 1.413 (8) Å, respectively]. A similar trend can be observed for the C—N bond lengths in (I) (Table 1).
In the crystal, the molecules of (I) are joined by a pair of nearly linear N—H···N hydrogen bonds into discrete dimers (Fig. 1 and Table 2). These interactions between the N-aryl-2-aminopyridine units assuming the E conformation generate a centrosymmetric R22(8) motif. As indicated above, the second N-aryl-2-aminopyridine unit adopts a nearly planar Z conformation and therefore cannot act as an H-atom acceptor in intermolecular hydrogen bonding. The NH group of this unit has no additional acceptor accessible and therefore is not involved in any hydrogen bonding.