Buy article online - an online subscription or single-article purchase is required to access this article.

The violet pigment methylbenzimidazolonodioxazine, C
22H
12Cl
2N
6O
4 (systematic name: 6,14-dichloro-3,11-dimethyl-1,3,9,11-tetrahydro-5,13-dioxa-7,15-diazadiimidazo-[4,5-
b:4′,5′-
m]pentacene-2,10-dione), shows an X-ray powder diagram consisting of only
ca 12 broad peaks. Indexing was not possible. The structure was solved by global lattice energy minimizations. The program
CRYSCA [Schmidt & Kalkhof (1999),
CRYSCA. Clariant GmbH, Pigments Research, Frankfurt am Main, Germany] was used to predict the possible crystal structures in different space groups. By comparing simulated and experimental powder diagrams, the correct structure was identified among the predicted structures. Owing to the low quality of the experimental powder diagram the Rietveld refinements gave no distinctive results and it was difficult to prove the correctness of the crystal structure. Finally, the structure was confirmed to be correct by refining the crystal structure of an isostructural mixed crystal having a better X-ray powder diagram. The compound crystallizes in
P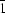
,
Z = 1. The crystal structure consists of a very dense packing of molecules, which are connected by hydrogen bridges of the type N—H

O=C. This packing explains the observed insolubility. The work shows that crystal structures of molecular compounds may be solved by lattice energy minimization from diffraction data of limited quality, even when indexing is not possible.
Supporting information
CCDC reference: 208781
Cell refinement: Fullprof for (I). Data reduction: Fullprof for (I). For both compounds, program(s) used to solve structure: CRYSCA (Energy minimization, M.U. Schmidt and H. Kalkhof, 1999). Program(s) used to refine structure: Fullprof for (I).
(I) 6,14-Dichlor-1,9-dimethyl-1,3,9,11-tetrahydro-diimidazo[4,5 − b:4',5'-m] triphendioxazin-2,10-dion
top
Crystal data top
| C22H12Cl2N6O4 | Z = 1 |
| Mr = 495.3 | F(000) = 252 |
| Triclinic, P1 | Unit cell determined by Rietveld refinement |
| a = 4.2753 (15) Å | Dx = 1.742 Mg m−3 |
| b = 8.311 (3) Å | Cu Kα radiation, λ = 1.54059 Å |
| c = 14.092 (5) Å | θ = 1.5–17° |
| α = 107.23 (3)° | T = 293 K |
| β = 93.53 (2)° | Powder, dark violet |
| γ = 97.17 (3)° | × × mm |
| V = 472.0 (3) Å3 | |
Data collection top
STOE Stadi-P, 0.7mm Capillary
diffractometer | 109 independent reflections |
| Radiation source: X-ray | θmax = 17°, θmin = 1.5° |
| Ge-111 monochromator | h = ?→? |
| Transmission scans | k = ?→? |
| 109 measured reflections | l = ?→? |
Refinement top
| wR(F2) = 0.212 | Primary atom site location: energy minimization |
| S = 11.5 | Secondary atom site location: energy minimization |
| 109 reflections | Hydrogen site location: energy minimization |
| 9 parameters | Calc |
| 3 restraints | |
Crystal data top
| C22H12Cl2N6O4 | β = 93.53 (2)° |
| Mr = 495.3 | γ = 97.17 (3)° |
| Triclinic, P1 | V = 472.0 (3) Å3 |
| a = 4.2753 (15) Å | Z = 1 |
| b = 8.311 (3) Å | Cu Kα radiation |
| c = 14.092 (5) Å | T = 293 K |
| α = 107.23 (3)° | × × mm |
Data collection top
STOE Stadi-P, 0.7mm Capillary
diffractometer | 109 independent reflections |
| 109 measured reflections | θmax = 17° |
Refinement top
| wR(F2) = 0.212 | 9 parameters |
| S = 11.5 | 3 restraints |
| 109 reflections | Calc |
Special details top
Experimental. Structure determined from X-ray powder data. Synchrotron data Structure solution: by energy minimization using the program CRYSCA (Martin U. Schmidt & Holger Kalkhof, 1999). Rietveld refinement: Program GSAS, 3 rigid bodies |
Geometry. Rigid body Rietveld refinement. Geometry of molecule taken from crystal structures of other compounds, and not further refined |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| C1 | 0.05795 (1) | −0.14104 (1) | 0.03130 (1) | 0.0* | |
| C2 | 0.23043 (1) | 0.02443 (1) | 0.08370 (1) | 0.0* | |
| C3 | 0.15843 (1) | 0.16338 (1) | 0.04733 (1) | 0.0* | |
| O4 | 0.31404 (1) | 0.32352 (1) | 0.09386 (1) | 0.0* | |
| C5 | 0.53528 (1) | 0.34715 (1) | 0.17420 (1) | 0.0* | |
| C6 | 0.59515 (1) | 0.20735 (1) | 0.20612 (1) | 0.0* | |
| N7 | 0.43886 (1) | 0.04392 (1) | 0.15959 (1) | 0.0* | |
| Cl8 | 0.13283 (1) | −0.31079 (1) | 0.07087 (1) | 0.0* | |
| C9 | 0.68490 (1) | 0.51019 (1) | 0.21830 (1) | 0.0* | |
| C10 | 0.90776 (1) | 0.53398 (1) | 0.29924 (1) | 0.0* | |
| C11 | 0.97645 (1) | 0.39784 (1) | 0.33418 (1) | 0.0* | |
| C12 | 0.81955 (1) | 0.23540 (1) | 0.28733 (1) | 0.0* | |
| N13 | 1.21072 (1) | 0.46936 (1) | 0.41602 (1) | 0.0* | |
| C14 | 1.27974 (1) | 0.64470 (1) | 0.42939 (1) | 0.0* | |
| O15 | 1.47337 (1) | 0.74718 (1) | 0.49401 (1) | 0.0* | |
| N16 | 1.09234 (1) | 0.68229 (1) | 0.35732 (1) | 0.0* | |
| C17 | 1.09366 (1) | 0.85003 (1) | 0.34613 (1) | 0.0* | |
| H18 | 0.63464 (1) | 0.61043 (1) | 0.19270 (1) | 0.0* | |
| H19 | 0.86889 (1) | 0.13472 (1) | 0.31263 (1) | 0.0* | |
| H20 | 1.31373 (1) | 0.40765 (1) | 0.45849 (1) | 0.0* | |
| H21 | 1.10320 (1) | 0.93834 (1) | 0.41628 (1) | 0.0* | |
| H22 | 0.89605 (1) | 0.84880 (1) | 0.29907 (1) | 0.0* | |
| H23 | 1.29311 (1) | 0.87929 (1) | 0.31237 (1) | 0.0* | |
Geometric parameters (Å, º) top
| C1—C2 | 1.4349 (1) | C10—N16 | 1.3756 (1) |
| C1—Cl8 | 1.7207 (1) | C11—C12 | 1.3785 (1) |
| C2—C3 | 1.4529 (1) | C11—N13 | 1.4106 (1) |
| C2—N7 | 1.3070 (1) | C12—H19 | 1.0408 (1) |
| C3—O4 | 1.3611 (1) | N13—C14 | 1.4034 (1) |
| O4—C5 | 1.3803 (1) | N13—H20 | 1.0104 (1) |
| C5—C6 | 1.4098 (1) | C14—O15 | 1.2296 (1) |
| C5—C9 | 1.3666 (1) | C14—N16 | 1.3861 (1) |
| C6—N7 | 1.3833 (1) | N16—C17 | 1.4481 (1) |
| C6—C12 | 1.3912 (1) | C17—H21 | 1.0370 (1) |
| C9—C10 | 1.3905 (1) | C17—H22 | 1.0385 (1) |
| C9—H18 | 1.0409 (1) | C17—H23 | 1.0389 (1) |
| C10—C11 | 1.4157 (1) | | |
| | | |
| C2—C1—Cl8 | 118.4468 (1) | C10—C11—N13 | 106.2138 (1) |
| C1—C2—C3 | 116.2728 (1) | C12—C11—N13 | 134.0398 (1) |
| C1—C2—N7 | 120.1458 (1) | C6—C12—C11 | 119.4672 (1) |
| C3—C2—N7 | 123.5814 (1) | C6—C12—H19 | 120.3846 (1) |
| C2—C3—O4 | 119.1306 (1) | C11—C12—H19 | 120.1482 (1) |
| C3—O4—C5 | 118.3039 (1) | C11—N13—C14 | 107.9976 (1) |
| O4—C5—C6 | 120.0153 (1) | C11—N13—H20 | 127.0901 (1) |
| O4—C5—C9 | 116.0361 (1) | C14—N13—H20 | 124.9123 (1) |
| C6—C5—C9 | 123.9486 (1) | N13—C14—O15 | 125.7657 (1) |
| C5—C6—N7 | 122.3299 (1) | N13—C14—N16 | 108.2848 (1) |
| C5—C6—C12 | 118.612 (1) | O15—C14—N16 | 125.9495 (1) |
| N7—C6—C12 | 119.0580 (1) | C10—N16—C14 | 108.3808 (1) |
| C2—N7—C6 | 116.6389 (1) | C10—N16—C17 | 126.4254 (1) |
| C5—C9—C10 | 116.0303 (1) | C14—N16—C17 | 125.1939 (1) |
| C5—C9—H18 | 121.8652 (1) | N16—C17—H21 | 108.8894 (1) |
| C10—C9—H18 | 122.1045 (1) | N16—C17—H22 | 109.3328 (1) |
| C9—C10—C11 | 122.1954 (1) | N16—C17—H23 | 108.8735 (1) |
| C9—C10—N16 | 128.6816 (1) | H21—C17—H22 | 112.9502 (1) |
| C11—C10—N16 | 109.1231 (1) | H21—C17—H23 | 109.2285 (1) |
| C10—C11—C12 | 119.7465 (1) | H22—C17—H23 | 107.4900 (1) |
(calc) 6,14-Dichlor-1,9-dimethyl-1,3,9,11-tetrahydro-diimidazo[4,5 − b:4',5'-m] triphendioxazin-2,10-dion
top
Crystal data top
| C22H12Cl2N6O4 | γ = 95.1180 (1)° |
| Mr = 495.3 | V = 481.93 (1) Å3 |
| Triclinic, P1 | Z = 1 |
| a = 4.3346 (1) Å | Crystal structure calculated by lattice energy minimization without
reference to experimental data. |
| b = 8.4193 (1) Å | Cu Kα radiation, λ = 1.54059 Å |
| c = 13.9057 (1) Å | T = 293 K |
| α = 106.9467 (1)° | Powder, dark violet |
| β = 92.9106 (1)° | × × mm |
Data collection top
STOE Stadi-P, 0.7mm Capillary
diffractometer | θmax = 17°, θmin = 1.5° |
| Radiation source: X-ray | h = ?→? |
| Ge-111 monochromator | k = ?→? |
| Transmission scans | l = ?→? |
Refinement top
| Primary atom site location: lattice energy minimization | Calc |
| Secondary atom site location: lattice energy minimization | |
| Hydrogen site location: lattice energy minimization | |
Crystal data top
| C22H12Cl2N6O4 | β = 92.9106 (1)° |
| Mr = 495.3 | γ = 95.1180 (1)° |
| Triclinic, P1 | V = 481.93 (1) Å3 |
| a = 4.3346 (1) Å | Z = 1 |
| b = 8.4193 (1) Å | Cu Kα radiation |
| c = 13.9057 (1) Å | T = 293 K |
| α = 106.9467 (1)° | × × mm |
Data collection top
STOE Stadi-P, 0.7mm Capillary
diffractometer | θmax = 17° |
Special details top
Experimental. Crystal structure calculated by lattice energy minimization using the program CRYSCA (Martin U. Schmidt & Holger Kalkhof, 1999). without any reference to experimental data. The simulated powder diagram of the calculated structure is similar to the experimental X-ray powder diagram (no fit). No Rietveld refinement. |
Geometry. Rigid molecule. Geometry of molecule taken from crystal structures of other compounds, and not further optimized |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| C1 | 0.06106 (1) | −0.13993 (1) | 0.03139 (1) | 0.0* | |
| C3 | 0.22942 (1) | 0.02065 (1) | 0.08267 (1) | 0.0* | |
| C2 | 0.15438 (1) | 0.15873 (1) | 0.04628 (1) | 0.0* | |
| O1 | 0.30602 (1) | 0.31430 (1) | 0.09177 (1) | 0.0* | |
| C5 | 0.52628 (1) | 0.33430 (1) | 0.17113 (1) | 0.0* | |
| C4 | 0.58923 (1) | 0.19557 (1) | 0.20313 (1) | 0.0* | |
| N1 | 0.43699 (1) | 0.03676 (1) | 0.15764 (1) | 0.0* | |
| Cl1 | 0.13968 (1) | −0.30841 (1) | 0.07105 (1) | 0.0* | |
| C6 | 0.67187 (1) | 0.49282 (1) | 0.21421 (1) | 0.0* | |
| C7 | 0.89375 (1) | 0.51295 (1) | 0.29416 (1) | 0.0* | |
| C8 | 0.96542 (1) | 0.37770 (1) | 0.32913 (1) | 0.0* | |
| C9 | 0.81254 (1) | 0.21988 (1) | 0.28333 (1) | 0.0* | |
| N10 | 1.19759 (1) | 0.44472 (1) | 0.40982 (1) | 0.0* | |
| C11 | 1.26246 (1) | 0.61657 (1) | 0.42248 (1) | 0.0* | |
| O11 | 1.45336 (1) | 0.71473 (1) | 0.48604 (1) | 0.0* | |
| N2 | 1.07457 (1) | 0.65643 (1) | 0.35111 (1) | 0.0* | |
| C16 | 1.07205 (1) | 0.82180 (1) | 0.33951 (1) | 0.0* | |
| H6 | 0.61942 (1) | 0.59240 (1) | 0.18858 (1) | 0.0* | |
| H9 | 0.86409 (1) | 0.11987 (1) | 0.30866 (1) | 0.0* | |
| H10 | 1.30181 (1) | 0.38234 (1) | 0.45200 (1) | 0.0* | |
| H161 | 1.08264 (1) | 0.90885 (1) | 0.41051 (1) | 0.0* | |
| H162 | 0.87597 (1) | 0.82358 (1) | 0.29368 (1) | 0.0* | |
| H163 | 1.26595 (1) | 0.84749 (1) | 0.30325 (1) | 0.0* | |
Experimental details
| | (I) | (calc) |
| Crystal data |
| Chemical formula | C22H12Cl2N6O4 | C22H12Cl2N6O4 |
| Mr | 495.3 | 495.3 |
| Crystal system, space group | Triclinic, P1 | Triclinic, P1 |
| Temperature (K) | 293 | 293 |
| a, b, c (Å) | 4.2753 (15), 8.311 (3), 14.092 (5) | 4.3346 (1), 8.4193 (1), 13.9057 (1) |
| α, β, γ (°) | 107.23 (3), 93.53 (2), 97.17 (3) | 106.9467 (1), 92.9106 (1), 95.1180 (1) |
| V (Å3) | 472.0 (3) | 481.93 (1) |
| Z | 1 | 1 |
| Radiation type | Cu Kα | Cu Kα |
| µ (mm−1) | ? | ? |
| Crystal size (mm) | × × | × × |
| |
| Data collection |
| Diffractometer | STOE Stadi-P, 0.7mm Capillary
diffractometer | STOE Stadi-P, 0.7mm Capillary
diffractometer |
| Absorption correction | – | – |
No. of measured, independent and
observed (?) reflections | 109, 109, ? | ?, ?, ? |
| Rint | ? | ? |
| θmax (°) | 17 | 17 |
| (sin θ/λ)max (Å−1) | 0.190 | 0.190 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | ?, 0.212, 11.5 | ?, ?, ? |
| No. of reflections | 109 | ? |
| No. of parameters | 9 | ? |
| No. of restraints | 3 | ? |
| H-atom treatment | Calc | Calc |
| Δρmax, Δρmin (e Å−3) | ?, ? | ?, ? |

 Subscribe to Acta Crystallographica Section B: Structural Science, Crystal Engineering and Materials
Subscribe to Acta Crystallographica Section B: Structural Science, Crystal Engineering and Materials
The full text of this article is available to subscribers to the journal.
If you have already registered and are using a computer listed in your registration details, please email
[email protected] for assistance.


 journal menu
journal menu research papers
research papers










![[Figure 1]](https://journals.iucr.org/b/issues/2005/01/00/av5015/av5015fig1thm.gif)
![[Figure 2]](https://journals.iucr.org/b/issues/2005/01/00/av5015/av5015fig2thm.gif)
![[Figure 3]](https://journals.iucr.org/b/issues/2005/01/00/av5015/av5015fig3thm.gif)
![[Figure 4]](https://journals.iucr.org/b/issues/2005/01/00/av5015/av5015fig4thm.gif)
![[Figure 5]](https://journals.iucr.org/b/issues/2005/01/00/av5015/av5015fig5thm.gif)
![[Figure 6]](https://journals.iucr.org/b/issues/2005/01/00/av5015/av5015fig6thm.gif)
![[Figure 7]](https://journals.iucr.org/b/issues/2005/01/00/av5015/av5015fig7thm.gif)



